(JO n° 297 du 23 décembre 1994)
Texte abrogé par l'article 1er de l'Arrêté du 24 août 2017 (JO n° 216 du 15 septembre 2017)
NOR : AGRG9402177A
Texte modifié par :
Décret n° 2010-429 du 29 avril 2010 (JO n° 101 du 30 avril 2010)
Décret n° 2006-1177 du 22 septembre 2006 (JO du 23 septembre 2006)
Arrêté 28 février 2005 (JO du 14 avril 2005)
Décret n° 2003-768 du 1er août 2003 (JO du 7 août 2003)
Arrêté 23 avril 2003 (JO du 29 mai 2003)
Arrêté du 17 décembre 2002 (JO du 14 janvier 2003)
Ordonnance n° 2000-914 du 18 septembre 2000 (JO du 21 septembre 2000)
Arrêté du 1er décembre 1998 (JO du 7 février 1998)
Arrêté du 1er février 1996 (JO du 20 février 1996)
Vus
Le ministre de l'économie, le ministre de l'industrie, des postes et télécommunications et du commerce extérieur, le ministre de l'agriculture et de la pêche et le ministre de l'environnement,
Vu la directive n° 91/414/C.E.E. du conseil du 15 juillet 1991 concernant la mise sur le marché des produits phytopharmaceutiques, dont les annexes ont été modifiées par la directive n° 93/71/C.E.E. de la commission du 27 juillet 1993 ;
Vu le règlement n° 3600/92 de la commission du 11 décembre 1992 établissant les modalités de mise en œuvre de la première phase du programme de travail visé à l'article 8, paragraphe 2, de la directive n° 91/414/C.E.E. ;
Vu le code de la santé publique, et notamment le chapitre Ier (Substances vénéneuses) du titre III (Restrictions au commerce de certaines substances et de certains objets) du livre V (Pharmacie), et notamment ses articles R. 5149 à R. 5170 ;
Vu le code de la consommation ;
Vu la loi du 4 août 1903 réglementant le commerce des produits anticryptogamiques ;
Vu la loi n° 43-525 du 2 novembre 1943 validée et modifiée relative à l'organisation du contrôle des produits antiparasitaires à usage agricole ;
Vu la loi n° 75-633 du 15 juillet 1975 modifiée relative à l'élimination des déchets et à la récupération des matériaux ;
Vu le décret du 11 mai 1937 portant règlement d'administration publique pour l'application de la loi du 4 août 1903, modifiée par la loi du 10 mars 1935, concernant la répression des fraudes dans le commerce des produits utilisés pour la destruction des ravageurs ;
Vu le décret n° 74-682 du 1er août 1974 modifié pris pour l'application de la loi du 2 novembre 1943 ;
Vu le décret n° 88-1231 du 29 décembre 1988 relatif à certaines substances et préparations dangereuses ;
Vu le décret n° 90-562 du 3 juillet 1990 modifiant le décret n° 78-838 du 2 août 1978 pris pour l'application de l'article 10 de la loi du 2 novembre 1943 modifiée autorisant la perception de droit de contrôle au titre de l'homologation des produits antiparasitaires à usage agricole ;
Vu le décret n° 94-359 du 5 mai 1994 relatif au contrôle des produits phytopharmaceutiques ;
Vu l'arrêté du 1er décembre 1987 relatif à l'homologation des produits visés à l'article 1er de la loi du 2 novembre 1943 ;
Vu l'arrêté du 28 mars 1989 fixant les conditions de classement, d'étiquetage et d'emballage des préparations pesticides ;
Vu l'arrêté du 28 mars 1989 fixant la liste et les conditions d'étiquetage et d'emballage des substances et préparations dangereuses ou vénéneuses ;
Vu l'arrêté du 20 avril 1994 relatif à la déclaration, la classification, l'emballage et l'étiquetage des substances ;
Vu l'avis de la commission des produits antiparasitaires à usage agricole et des produits assimilés,
Titre Ier : Dispositions relatives à l'expérimentation des produits phytopharmaceutiques
Section 1 : L'autorisation de distribution pour expérimentation
Article 1er de l'arrêté du 6 septembre 1994
(Décret n° 2010-429 du 29 avril 2010, article 6 V)
Pour être testés ou expérimentés, les produits phytopharmaceutiques qui n'ont pas déjà bénéficié d'une autorisation de mise sur le marché doivent obtenir au préalable une autorisation de distribution pour expérimentation.
Au sens du présent arrêté, on entend par :
Essais de recherche : les essais qui permettent de définir le champ d'action de la substance active et du produit, et / ou de sélectionner le ou les usages agronomiques qui feront l'objet d'une demande d'autorisation de mise sur le marché. Ces essais sont réalisés dans des installations de la société qui développe le produit ou sur les installations qui sont sous son contrôle ;
Essais d'homologation : les essais réalisés par des instances officielles ( directions régionales de l'alimentation, de l'agriculture et de la forêt, Institut national de la recherche agronomique) ou structures agréées " Bonnes pratiques d'expérimentation ou Bonnes pratiques de laboratoire " (firmes, instituts, distributeurs, prestataires de service) qui permettent d'acquérir les données qui constituent la partie efficacité du dossier d'autorisation de mise sur le marché ;
Essais de connaissance régionale : les essais conduits chez des prescripteurs, ou chez un agriculteur, sous la responsabilité de la société qui développe le produit et dépose le dossier d'autorisation de mise sur le marché, qui ont pour objectif de valider les préconisations d'emploi du produit et de procéder à son référencement commercial.
Le délai maximal entre le dépôt de la demande d'autorisation de distribution pour expérimentation et la décision ne pourra excéder six mois.
Par extension et seulement pour les essais de connaissance régionale, un produit phytopharmaceutique ayant déjà obtenu une autorisation de mise sur le marché pour d'autres groupes de cultures que celles faisant l'objet du test ou de l'expérimentation doit bénéficier d'une autorisation préalable de distribution pour expérimentation pour cette nouvelle culture.
Toutefois, les exigences prévues aux premier, sixième et septième alinéas précédents ne sont pas requises pour les essais de recherche effectuées par des personnes travaillant dans les laboratoires, stations de recherche et domaines expérimentaux publics ou privés, reconnus par l'Etat.
Article 2 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, articles 1er et 3)
I. La demande d'autorisation de distribution pour expérimentation du produit phytopharmaceutique doit être adressée au ministère chargé de l'agriculture (direction générale de l'alimentation, sous-direction de la protection des végétaux) au moins six mois avant le début de l'expérimentation par le signataire de ladite demande, responsable de la distribution pour expérimentation.
II. Le demandeur doit fournir toutes les informations dont il dispose concernant l'efficacité et l'innocuité du produit ou, à défaut, au moins les informations disponibles permettant d'évaluer les effets éventuels sur la santé et sur l'environnement.
III. La demande d'autorisation de distribution pour expérimentation doit comprendre :
- un formulaire de demande, établi en trois exemplaires ;
- un dossier, établi en trois exemplaires, contenant l'ensemble des informations visées à l'alinéa II, ci-dessus.
Article 3 de l'arrêté du 6 septembre 1994
(Décret n° 2003-768 du 1er août 2003, article 3 V)
L'autorisation de distribution pour expérimentation d'un produit phytopharmaceutique est délivrée par le ministre chargé de l'agriculture conformément à la procédure prévue par l'article R. 253-21 du code rural.
Elle est accordée pour des quantités et des zones limitées, pour une durée n'excédant pas deux ans et sous réserve qu'à chaque test une déclaration d'expérimentation soit effectuée selon les modalités prévues aux articles 7 à 7-2 du présent arrêté. Elle est renouvelable, dans les mêmes conditions.
Article 4 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, article 1er)
Les emballages ou contenants des produits phytopharmaceutiques bénéficiant d'une autorisation de distribution pour expérimentation, mis à la disposition des expérimentateurs, doivent porter de façon apparente, lisible et en caractères indélébiles, les indications suivantes :
a) Le nom de fantaisie du produit ;
b) Le numéro de l'autorisation de distribution pour expérimentation ;
c) Le nom, l'adresse et le numéro de téléphone du détenteur de l'autorisation de distribution pour expérimentation ;
d) Les phrases types indiquant la nature des risques particuliers pour l'homme, les animaux ou l'environnement ;
e) Les phrases types de précaution à prendre pour la protection de l'homme, des animaux ou de l'environnement ;
f) Les autres précautions d'emploi ou contre-indications, figurant le cas échéant sur la décision d'autorisation de distribution pour expérimentation ;
g) Les instructions d'emploi et la dose à appliquer par culture ou type de culture ;
h) La mention spécifique "produit pour usage expérimental seulement".
Article 5 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, article 1er)
Les produits phytopharmaceutiques bénéficiant d'une autorisation de distribution pour expérimentation doivent être testés ou expérimentés dans les conditions d'emploi prescrites mentionnées sur l'étiquette.
A l'issue de la campagne d'expérimentation réalisée dans le cadre des essais de connaissance régionale, le bénéficiaire de l'autorisation de distribution pour expérimentation doit fournir au ministre chargé de l'agriculture un rapport succinct faisant une présentation synthétique des résultats de ces essais.
Article 6 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, article 1er)
Toute publicité concernant les produits phytopharmaceutiques bénéficiant d'une autorisation de distribution pour expérimentation est interdite.
Section 2 : La déclaration d'expérimentation
Article 7 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, articles 1er et 6)
Tout test ou expérimentation d'un produit phytosanitaire conduit dans le cadre du dispositif général d'autorisation de distribution pour expérimentation doit faire l'objet d'une déclaration, sous la responsabilité du demandeur, auprès du ministre chargé de l'agriculture.
Article 7-1 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, articles 1er et 7)
La déclaration doit contenir les informations suivantes :
1. La référence (ou numéro) de l'autorisation de distribution d'expérimentation à laquelle se rattache le test ou l'expérimentation (si nécessaire) ;
2. Les coordonnées de l'organisme réalisant le test ou l'expérimentation (nom, raison sociale, adresse, téléphone, télécopie, adresse électronique, numéro d'agrément pour les essais d'homologation) ;
3. Les renseignements utiles sur le produit et sur la société détentrice du produit, tels que le nom du produit, numéro d'autorisation de mise sur le marché du produit (si disponibles) ;
4. Les renseignements sur le test ou l'expérimentation : culture, localisation du test (nom et code postal de la commune).
Article 7-2 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, article 8)
La déclaration doit être effectuée auprès du ministre chargé de l'agriculture au plus tard un mois après la mise en place du test ou de l'expérimentation concernés.
Section 3 : Le devenir des récoltes au terme de l'expérimentation
Article 8 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, articles 1er et 9)
A l'issue de toute expérimentation de produits phytopharmaceutiques, sauf dans le cas où le produit est utilisé conformément au respect des conditions d'emploi d'une autorisation déjà accordée pour la même culture permettant de respecter les limites maximales de résidus fixées, les produits récoltés utilisables à des fins alimentaires pour l'homme ou les animaux doivent être détruits.
Les produits sont éliminés dans les conditions prévues au titre IV du livre V du code de l'environnement.
Le détenteur de l'autorisation de distribution pour expérimentation doit fournir au ministre chargé de l'agriculture une attestation sur l'honneur de cette destruction précisant le lieu et le jour de sa réalisation.
Article 9 de l'arrêté du 6 septembre 1994
(Décret n° 2006-1177 du 22 septembre 2006, article 12 V)
Toute personne peut effectuer auprès du ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux) une demande motivée de dérogation à l'article 8 du présent arrêté. Si l'Agence française de sécurité sanitaire des aliments estime que les résidus du produit phytopharmaceutique testé ou expérimenté, dans le produit récolté, ne présentent pas de risques inacceptables, ces produits peuvent ne pas être détruits.
L'autorisation de ne pas détruire les produits récoltés est notifiée au demandeur par le ministre chargé de l'agriculture, sur proposition du comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés.
Article 9-1 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, articles 1er et 11)
Les produits expérimentés dans le cadre du présent titre sont soumis aux exigences prévues au titre III du présent arrêté.
Article 9-2 de l'arrêté du 6 septembre 1994
(Arrêté 23 avril 2003, articles 1er et 12)
Préalablement à toute décision de refus ou de retrait de l'autorisation de distribution pour expérimentation du produit phytopharmaceutique, le demandeur ou le détenteur de l'autorisation disposent d'un délai qui leur est notifié par le ministre chargé de l'agriculture pour présenter leurs observations et se mettre en conformité avec les exigences requises.
Titre II : L'autorisation de mise sur le marché des produits phytopharmaceutiques.
Article 10 de l'arrêté du 6 septembre 1994
L'autorisation de mise sur le marché d'un produit phytopharmaceutique est octroyée pour un ou plusieurs usages demandés :
a) Si ses substances actives sont inscrites sur la liste communautaire des substances actives ;
b) Et si le produit révèle, dans les conditions d'emploi prescrites, son efficacité, sa sélectivité à l'égard des végétaux et des produits végétaux et son innocuité à l'égard de la santé humaine, animale et de l'environnement.
Article 11 de l'arrêté du 6 septembre 1994
Par dérogation au a de l'article 10 ci-dessus, l'autorisation de mise sur le marché peut être délivrée :
a) Lorsque la substance active est en cours d'inscription selon la procédure définie à l'article 12 du présent arrêté et si les dossiers déposés par le demandeur de l'inscription ont été jugés conformes par les instances communautaires concernées selon la procédure définie à l'article 13 du présent arrêté ;
b) Lorsque la substance active doit être révisée selon la procédure prévue à l'article 14 du présent arrêté.
Section 1 : Inscription de la substance active
Article 12 de l'arrêté du 6 septembre 1994
(Décret n° 2006-1177 du 22 septembre 2006, article 12 V)
I. Toute substance active qui n'entre pas dans la composition d'un produit phytopharmaceutique bénéficiant d'une autorisation de mise sur le marché avant le 25 juillet 1993 dans au moins un Etat membre de la Communauté européenne doit faire l'objet d'une inscription sur la liste communautaire des substances actives.
II. La demande d'inscription de la substance active est adressée au ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux) et doit comprendre :
- un dossier conforme à l'annexe I du présent arrêté concernant la substance active établi en trois exemplaires, ainsi que soixante-cinq résumés dudit dossier ;
- un dossier conforme à l'annexe II du présent arrêté concernant au moins un produit phytopharmaceutique contenant cette substance active établi en trois exemplaires, ainsi que soixante-cinq résumés dudit dossier.
III. La conformité des dossiers est notifiée par le ministre de l'agriculture et de la pêche au demandeur, après avis de l'Agence française de sécurité sanitaire des aliments et sur proposition du Comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés.
Si ces dossiers sont jugés conformes, le demandeur doit transmettre sa demande d'inscription de la substance active dans les meilleurs délais à la commission des communautés européennes et aux autres Etats membres.
Article 13 de l'arrêté du 6 septembre 1994
(Décret n° 2003-768 du 1er août 2003, article 3 V)
La demande d'inscription de la substance active est évaluée au niveau communautaire, conformément à la procédure prévue à l'article R. 253-10 I du code rural.
A l'issue de cette instruction, l'inscription est accordée par la Commission des communautés européennes pour une période renouvelable n'excédant pas dix ans.
Article 14 de l'arrêté du 6 septembre 1994
Toute substance active qui entre dans la composition d'un produit phytopharmaceutique bénéficiant d'une autorisation de mise sur le marché avant le 25 juillet 1993 dans au moins un Etat membre de la Communauté européenne doit faire l'objet d'une demande d'inscription auprès de la Commission des communautés européennes selon la procédure définie par le règlement n° 3600/92 de la commission du 11 décembre 1992 susvisé.
Section 2 : Régime de l'autorisation de mise sur le marché des produits phytopharmaceutiques
Article 15 de l'arrêté du 6 septembre 1994
La demande d'autorisation de mise sur le marché d'un produit phytopharmaceutique est adressée au ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux) et doit comprendre :
1. Un formulaire établi en trois exemplaires ;
2. Toute pièce d'ordre administratif nécessaire à l'instruction de la demande, établie en trois exemplaires ;
3. Une déclaration attestant que chacune des substances actives contenues dans le produit phytopharmaceutique est inscrite sur la liste communautaire des substances actives ;
4. Un dossier conforme à l'annexe II du présent arrêté, concernant le produit phytopharmaceutique, établi en trois exemplaires, ainsi que soixante-cinq résumés dudit dossier.
La demande d'autorisation de mise sur le marché est établie pour un produit phytopharmaceutique et pour un ou plusieurs usages.
Article 16 de l'arrêté du 6 septembre 1994
(Décret n° 2006-1177 du 22 septembre 2006, article 12 V)
I. La demande d'autorisation de mise sur le marché est instruite par l'Agence française de sécurité sanitaire des aliments et par le comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés.
L'évaluation des dossiers est réalisée en application des principes uniformes fixés en annexe III du présent arrêté.
II. Lors de l'instruction de la demande d'autorisation de mise sur le marché, le ministre de l'agriculture et de la pêche peut exiger toutes informations complémentaires qu'il juge nécessaires.
III. Lors de l'instruction, le comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés apprécie la compatibilité entre les usages demandés.
Article 17 de l'arrêté du 6 septembre 1994
Le ministre de l'agriculture et de la pêche prononce l'une des mesures suivantes concernant le produit phytopharmaceutique pour chacun des usages demandés :
a) L'autorisation de mise sur le marché pour une période n'excédant pas dix ans ;
b) Le maintien en étude sans autorisation de mise sur le marché ;
c) Le refus de l'autorisation de mise sur le marché ;
d) Le retrait de l'autorisation de mise sur le marché.
Le ministre de l'agriculture et de la pêche notifie l'une de ces mesures pour chacun des usages demandés sous forme d'un document officiel dénommé "décision d'autorisation" contresigné par celui-ci ou, par délégation, par son représentant.
Article 18 de l'arrêté du 6 septembre 1994
(Décret n° 2006-1177 du 22 septembre 2006, article 12 V)
En application du a de l'article 17 ci-dessus, l'autorisation est délivrée par le ministre de l'agriculture et de la pêche pour :
a) Dix ans lorsque l'ensemble des conditions exigées sont remplies ;
b) Une période n'excédant pas trois ans, lorsque la substance est en cours d'inscription en application du a de l'article 11 du présent arrêté ; cette période pouvant être prolongée lorsque aucune décision communautaire concernant la substance active n'est intervenue dans ce délai ;
c) Une période n'excédant pas quatre ans, lorsque des informations complémentaires sont demandées par le ministre de l'agriculture et de la pêche sur proposition du comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés et/ou après avis de l'Agence française de sécurité sanitaire des aliments ; cette période peut être prolongée, à titre exceptionnel, pour un délai maximum de deux ans.
La durée des autorisations de mise sur la marché visées aux b et c est applicable sans préjudice de la décision communautaire concernant l'inscription de la ou des substances actives contenues dans le produit.
Article 19 de l'arrêté du 6 septembre 1994
Lors de la première autorisation de mise sur le marché d'un produit phytopharmaceutique, le ministre de l'agriculture et de la pêche délivre un numéro d'autorisation.
Article 20 de l'arrêté du 6 septembre 1994
Le renouvellement de l'autorisation de mise sur le marché porte sur l'ensemble des usages pour lesquels le produit phytopharmaceutique a été autorisé. Elle intervient à la date d'expiration de sa validité, fixée au 31 décembre de la dixième année suivant celle au cours de laquelle la première autorisation a été accordée.
Article 21 de l'arrêté du 6 septembre 1994
Lorsque le produit phytopharmaceutique fait l'objet d'un maintien en étude sans autorisation de mise sur le marché pour un ou plusieurs usages demandés, le demandeur dispose d'un délai maximum de deux ans pour fournir les informations complémentaires nécessaires à la poursuite de l'instruction de la demande d'autorisation. Le ministre de l'agriculture et de la pêche peut prolonger, à titre exceptionnel, le délai de deux ans nécessaire à la fourniture des informations.
Article 22 de l'arrêté du 6 septembre 1994
Lorsqu'une autorisation de mise sur le marché est octroyée dans les conditions prévues à l'article 18 ou lorsqu'un maintien en étude sans autorisation de mise sur le marché est prononcé, le ministre de l'agriculture et de la pêche peut, sur proposition du comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés, demander la mise en place d'essais de contrôle. Ces essais sont réalisés par des services et organismes publics représentés au comité d'homologation des produits antiparasitaires à usage agricole et des produits assimilés.
Article 23 de l'arrêté du 6 septembre 1994
Au terme des délais visés aux articles 18 et 21 du présent arrêté, si les informations demandées ne sont pas fournies, le ministre de l'agriculture et de la pêche prononce soit le refus, soit le retrait de l'autorisation de mise sur le marché.
Article 24 de l'arrêté du 6 septembre 1994
Le demandeur ou le détenteur de l'autorisation de mise sur le marché qui entend retirer sa demande d'autorisation de mise sur le marché ou abandonner l'exploitation de son produit pour certains ou pour l'ensemble des usages pour lesquels le produit a été autorisé doit le notifier auprès du ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux).
Article 25 de l'arrêté du 6 septembre 1994
Tout changement de nom de l'organisme responsable de la demande d'autorisation de mise sur le marché d'un produit phytopharmaceutique ou de celui du détenteur de l'autorisation de mise sur le marché entraîne le dépôt d'une demande de transfert d'autorisation de mise sur le marché auprès du ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux).
Cette demande est accompagnée de toutes pièces administratives dûment remplies, demandées par le ministre de l'agriculture et de la pêche.
Article 26 de l'arrêté du 6 septembre 1994
Le détenteur de l'autorisation de mise sur le marché d'un produit phytopharmaceutique qui entend changer la dénomination commerciale du produit doit adresser au ministre de l'agriculture et de la pêche une nouvelle demande d'autorisation de mise sur le marché.
La demande doit faire mention de la nouvelle dénomination commerciale du produit et doit être conforme aux exigences mentionnées à l'article 15 du présent arrêté.
Article 27 de l'arrêté du 6 septembre 1994
Le détenteur ou le demandeur de l'autorisation de mise sur le marché qui entend changer l'une des informations fournies dans les formulaires de demande d'autorisation ou dans les pièces administratives l'accompagnant, ou qui souhaite modifier les conditions d'emploi du produit, doit en effectuer la demande auprès du ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux).
Article 28 de l'arrêté du 6 septembre 1994
Tout fait nouveau dans les informations fournies dans les dossiers conformes aux annexes I et II du présent arrêté par le demandeur ou le détenteur de l'autorisation doit être porté à la connaissance du ministère de l'agriculture et de la pêche (direction générale de l'alimentation, sous-direction de la protection des végétaux) et peut entraîner le réexamen de la demande d'autorisation de mise sur le marché.
Article 29 de l'arrêté du 6 septembre 1994
Le défaut de communication de fait nouveau dans les informations fournies par le demandeur ou le détenteur de l'autorisation de mise sur le marché peut entraîner le refus ou le retrait de celle-ci.
Article 30 de l'arrêté du 6 septembre 1994
Préalablement à toute décision de refus ou de retrait de l'autorisation de mise sur le marché du produit phytopharmaceutique, le demandeur ou le détenteur de l'autorisation disposent d'un délai qui leur est notifié par le ministre de l'agriculture et de la pêche pour présenter leurs observations et se mettre en conformité avec les exigences requises.
Titre III : L'emballage et l'étiquetage des produits mentionnés à l'article 1er de la loi du 2 novembre 1943.
Article 31 de l'arrêté du 6 septembre 1994
(Ordonnance n°2000-914 du 18 septembre 2000, article 11)
Sans préjudice des autres dispositions applicables en la matière, et notamment celles relatives aux substances et préparations dangereuses ou vénéneuses, les emballages ou contenants des produits définis à l'article L. 253-1 du code rural doivent répondre aux dispositions du présent titre.
Article 32 de l'arrêté du 6 septembre 1994
L'étiquette ou l'inscription doit être apposée de manière très apparente, lisible horizontalement lorsque l'emballage est en position normale.
L'étiquette doit adhérer par toute sa surface à l'emballage contenant directement la substance.
Si le produit est contenu dans plusieurs emballages, l'étiquette ou l'inscription doit figurer sur chacun d'eux.
Article 33 de l'arrêté du 6 septembre 1994
Sauf dispositions contraires, lorsqu'un produit fait l'objet d'une modification d'autorisation, un délai d'un an au plus à compter de la date de notification de la nouvelle décision est accordé au demandeur de la mise sur le marché pour mettre l'étiquetage en conformité.
A l'expiration de ce délai, un délai supplémentaire d'un an est toléré pour l'écoulement des stocks de ce produit sous l'ancien étiquetage par toute autre personne que le demandeur responsable de la mise sur le marché.
Article 34 de l'arrêté du 6 septembre 1994
(Arrêté du 28 février 2005, article 2)
Tout emballage ou contenant doit porter, de manière lisible et indélébile, les indications suivantes :
a) Le nom commercial du produit ;
b) Le nom et l'adresse du détenteur de l'autorisation ainsi que le numéro de l'autorisation du produit et, s'ils sont différents, le nom et l'adresse de la personne responsable de l'emballage et de l'étiquetage final ou de l'étiquetage final du produit sur le marché ;
c) Le nom et la quantité de chaque substance active exprimée :
- en p. 100 du poids pour les produits qui sont des produits solides, des aérosols, des liquides volatils (point d'ébullition maximale 50° C) ou visqueux (limite inférieure 1 Pa.s à 20° C) ;
- en p. 100 du poids et en gramme par litre à 2° C pour les autres liquides ;
- en p. 100 du volume pour les gaz ;
Le nom indiqué doit être celui figurant dans la décision d'autorisation, en conformité avec la nomenclature de la liste reprise à l'annexe I de l'arrêté du 20 avril 1994 susvisé ou, à défaut, son nom commun I.S.O. : si ce dernier nom n'existe pas, la substance active doit être désignée par sa désignation chimique selon la nomenclature U.I.C.P.A. ;
d) La quantité nette de produit indiquée en unité légale de mesure ;
e) Le numéro du lot de la préparation ou une indication permettant de l'identifier ;
f) L'indication de la nature des risques particuliers pour l'homme, les animaux ou l'environnement, sous forme de phrases types, choisies de manière appropriée conformément à l'annexe IV du présent arrêté ;
g) Les précautions à prendre pour la protection de l'homme, des animaux ou de l'environnement, sous forme de phrases types, choisies de manière appropriée conformément à l'annexe V du présent arrêté ;
h) Les autres précautions d'emploi ou contre-indications figurant le cas échéant sur la décision d'autorisation ;
i) Le type d'action exercée par le produit, par exemple insecticide, régulateur de croissance, herbicide, etc. ;
j) Le type de préparation, par exemple, poudre mouillable, concentré émulsionnable, etc ;
k) Les usages pour lesquels le produit est autorisé et les conditions spécifiques, notamment agricoles, phytosanitaires et environnementales, dans lesquelles le produit peut être utilisé ou doit, au contraire, être exclu ;
l) Les instructions d'emploi et la dose à appliquer pour chaque usage autorisé, exprimée en unités métriques ;
m) Si nécessaire, l'intervalle de sécurité à respecter pour chaque usage entre l'application et :
- le semis ou la plantation de la culture à protéger ;
- le semis ou la plantation des cultures ultérieures ;
- l'accès de l'homme ou des animaux à la culture traitée ;
- la récolte ;
- l'usage ou la consommation ;
n) Si nécessaire, les indications concernant la phytotoxicité éventuelle, la sensibilité variétale et tout autre effet secondaire direct ou indirect défavorable sur les produits végétaux ou les produits d'origine végétale, ainsi que les intervalles à observer entre l'application et le semis ou la plantation :
- de la culture concernée ;
- ou des cultures ultérieures ;
o) Des instructions pour l'élimination en toute sécurité du produit et de son emballage ;
p) La date de péremption dans des conditions normales de conservation, lorsque la durée de conservation du produit est limitée à moins de deux ans ;
q) La mention "réservé à un usage exclusivement professionnel" ou "autorisé pour l'emploi en jardins d'amateurs", ou autre, conformément à la décision d'autorisation de mise sur le marché.
Article 35 de l'arrêté du 6 septembre 1994
Si l'emballage ou le contenant est de dimension réduite, les indications requises aux points m, n, o, de l'article 34 ci-dessus peuvent être mentionnées sur une notice jointe à l'emballage.
Dans ce cas, l'emballage ou le contenant doivent porter la phrase : "Lire les instructions ci-jointes avant l'emploi".
Article 36 de l'arrêté du 6 septembre 1994
Lorsqu'un emballage ou un contenant contient des petits conditionnements prêts à l'emploi qui ne sont pas destinés à être vendus séparément, ceux-ci peuvent ne comporter que les mentions suivantes :
a) Le nom commercial ou désignation du produit ;
b) Nom et adresse du demandeur responsable de la mise sur le marché ;
c) Symboles et indications de danger le cas échéant ;
d) Le nom des substances actives et des substances dangereuses prévues par la réglementation en vigueur.
Article 37 de l'arrêté du 6 septembre 1994
Lorsqu'un emballage ou un contenant contient des sachets hydrosolubles, ceux-ci doivent porter au moins les indications suivantes :
a) Le nom commercial ou désignation du produit ;
b) Sachet hydrosoluble ou soluble :
A conserver dans l'emballage d'origine à l'abri de l'humidité ;
Se référer aux conditions et précautions d'emploi mentionnées sur l'emballage.
Dans ce cas, l'emballage ou le contenant doit porter des indications appropriées sur le mode d'emploi des sachets hydrosolubles.
Titre IV : Dispositions modifiant l'arrêté du 1er décembre 1987 susvisé
Article 38 de l'arrêté du 6 septembre 1994
Les dispositions de l'arrêté du 1er décembre 1987 susvisé qui concernent les produits phytopharmaceutiques tels que définis à l'article 1er du décret n° 94-359 du 5 mai 1994 sont abrogées.
Article 39 de l'arrêté du 6 septembre 1994
I. Les termes " produits visés à l'article 1er de la loi du 2 novembre 1943 " du titre de l'arrêté du 1er décembre 1987 susvisé sont remplacés par " produits visés aux points 4 et 7 de l'article 1er de la loi du 2 novembre 1943 ".
II. Dans l'article 1er de l'arrêté du 1er décembre 1987 susvisé, les termes " produits énumérés à l'article 1er de la loi du 2 novembre 1943 " sont remplacés par les termes " produits énumérés aux points 4 et 7 de l'article 1er de la loi du 2 novembre 1943 ".
Article 40 de l'arrêté du 6 septembre 1994
Les dispositions de l'article 9 de l'arrêté du 1er décembre 1987 précité sont remplacées comme suit:
" Préalablement à toute décision de refus ou de retrait d'homologation d'une spécialité, le demandeur ou le détenteur de l'homologation dispose d'un délai qui lui est notifié par le ministre de l'agriculture et de la pêche pour présenter ses observations et se mettre en conformité avec les exigences requises. "
Article 41 de l'arrêté du 6 septembre 1994
Le directeur général de l'alimentation, le directeur général des stratégies industrielles, le directeur général de la concurrence, de la consommation et de la répression des fraudes et le directeur de la prévention de la pollution et des risques sont chargés, chacun en ce qui le concerne, de l'exécution du présent arrêté.
Le ministre de l'agriculture et de la pêche,
Pour le ministre et par délégation :
Le directeur général de l'alimentation,
P. Guerin.
Le ministre de l'économie,
Pour le ministre et par délégation :
Le directeur général de la concurrence, de la consommation et de la répression des fraudes,
C. Babusiaux.
Le ministre de l'industrie, des postes et télécommunications et du commerce extérieur,
Pour le ministre et par délégation :
Le directeur général des stratégies industrielles,
D. Lombard.
Le ministre de l'environnement,
Pour le ministre et par délégation :
Le directeur de la prévention de la pollution et des risques,
G. Defrance.
Annexe I : Conditions à remplir pour introduire le dossier d'insertion d'une substance active dans la liste communautaire des substances actives
Introduction
(Arrêté du 1er février 1996, articles 3 et 4)
L'information doit :
1.1. Comprendre un dossier technique fournissant les informations nécessaires pour évaluer les risques prévisibles, immédiats ou à plus long terme, que la substance peut comporter pour l'homme, les animaux et l'environnement et contenant au moins les résultats des études visées ci-après ;
1.2. Le cas échéant, être recueillie conformément à la version la plus récente des lignes directrices, visées ou décrites dans la présente annexe ; pour les études commencées avant la mise en vigueur de la modification de la présente annexe, l'information doit être recueillie conformément à des lignes directrices adéquates, validées à l'échelon national ou international, ou, en leur absence, à des lignes directrices acceptées par l'autorité compétente ;
1.3. Comprendre, si la ligne directrice ne convient pas ou n'est pas décrite, ou si l'on a utilisé une autre ligne directrice que celles qui sont visées dans la présente annexe, une justification de la ligne directrice utilisée qui soit acceptable pour l'autorité compétente ;
En particulier lorsqu'il est fait référence dans cette annexe à une méthode CE qui est la transposition d'une méthode mise au point par une organisation internationale (par exemple l'O.C.D.E.), les Etats membres peuvent accepter que l'information requise soit recueillie conformément à la version la plus récente de cette méthode si, au début des études, la méthode CE n'a pas encore été mise à jour ;
1.4. Comprendre, si l'autorité compétente l'exige, une description complète des lignes directrices utilisées, à moins qu'il n'y soit fait référence ou qu'elles soient décrites dans la présente annexe, ainsi qu'une description complète de toute variante ainsi que sa justification, acceptable pour l'autorité compétente ;
1.5. Comprendre un rapport complet et impartial des études menées ainsi que leur description complète ou une justification acceptable pour l'autorité compétente lorsque :
- certaines données ou informations particulières qui ne semblent pas nécessaires en raison de la nature de la substance ou des utilisations qui en sont proposées ne sont pas fournies,
ou
- il n'est pas scientifiquement nécessaire ou techniquement possible de fournir les informations et les données ;
1.6. Le cas échéant, avoir été recueillie conformément aux dispositions de la directive n° 86/609/C.E.E.
(Arrêté du 17 décembre 2002, article 1er)
2.1. Les essais et analyses doivent être effectués conformément aux principes fixés dans la directive n° 87/18/C.E.E., lorsqu'ils ont pour but de recueillir des données sur les propriétés intéressant la santé humaine et animale ou l'environnement et/ou sur la sécurité dans ces domaines.
2.2. Par dérogation au point 2.1, les Etats membres peuvent prévoir que les essais et les analyses effectués sur leur territoire, visant à recueillir des données sur les propriétés et/ou la sécurité des substances en ce qui concerne les abeilles et les arthropodes utiles autres que les abeilles, seront confiés à des services ou à des organismes d'essais officiels ou officiellement reconnus remplissant au moins les conditions stipulées aux points 2.2 et 2.3 de l'introduction de l'annexe II.
Cette dérogation s'applique aux essais qui ont effectivement débuté au plus tard le 31 décembre 1999.
2.3. Par dérogation au point 2.1, les Etats membres peuvent prévoir que les essais contrôlés sur les résidus, effectués sur leur territoire conformément aux dispositions de la section 6 Résidus dans ou sur les produits, la nourriture et l'alimentation traités, avec des produits phytosanitaires contenant les substances actives déjà présentes sur le marché deux ans après la notification de la directive, seront réalisés par des services ou des organismes d'essais officiels ou officiellement reconnus, remplissant au minimum les conditions visées aux points 2.2 et 2.3 de l'introduction de l'annexe II.
Cette dérogation s'applique aux essais contrôlés sur les résidus ayant effectivement débuté au plus tard le 31 décembre 1997.
2.4. Par dérogation au point 2.1, pour les substances actives constituées de micro-organismes ou de virus, les tests et les analyses effectués afin de recueillir des données sur les propriétés et/ou la sécurité en ce qui concerne des aspects autres que la santé humaine peuvent être réalisés par des services ou des organismes d'expérimentation officiels ou officiellement reconnus remplissant au minimum les conditions visées aux points 2.2 et 2.3 de l'introduction de l'annexe II.
Partie A : Substances.
(Arrêté 1er février 1996, article 1er)
On entend par substance les éléments chimiques et leurs composés tels qu'ils se présentent à l'état naturel ou tels que produits par l'industrie, incluant toute impureté résultant inévitablement du procédé de fabrication.
1. Identité de la substance active
L'information fournie doit permettre d'identifier chaque substance active avec précision, d'en définir la spécification et d'en caractériser la nature. Ces données et informations sont requises pour toutes les substances actives, sauf indication contraire.
1.1. Demandeur (nom, adresse, etc.).
Indiquer le nom et l'adresse du demandeur (adresse permanente dans la Communauté), ainsi que le nom, la position, le numéro de téléphone et de télécopieur de la personne à contacter.
Lorsque, en outre, le demandeur a un bureau, un agent ou un représentant dans l'Etat membre auquel la demande d'insertion est présentée et, s'il est différent, dans l'Etat membre rapporteur nommé par la Commission, indiquer le nom et l'adresse du bureau, de l'agent ou du représentant local, ainsi que le nom, la position, le numéro de téléphone et de télécopieur de la personne à contacter.
1.2. Fabricant (nom, adresse, y compris l'emplacement de l'installation).
Indiquer le nom et l'adresse du ou des fabricants de la substance active, ainsi que le nom et l'adresse de chaque installation dans laquelle la substance active est fabriquée. Indiquer un point de contact (de préférence central, avec nom, numéro de téléphone et de télécopieur), auquel seront envoyées les informations d'actualisation et où il sera répondu aux questions qui se posent au sujet de la technologie de fabrication, des procédés et de la qualité du produit (y compris, le cas échéant, au sujet des lots individuels).
Si l'emplacement ou le nombre des fabricants est modifié après l'insertion de la substance active, notifier de nouveau l'information requise à la Commission et aux Etats membres.
1.3. Nom commun proposé ou accepté par l'ISO (Organisation de normalisation internationale) et synonymes.
Indiquer le nom commun ISO ou proposé par l'ISO et, le cas échéant, d'autres noms communs proposés ou acceptés (synonymes), y compris le nom (titre) de l'autorité responsable de la nomenclature concernée.
1.4. Dénomination chimique (nomenclature de l'UICPA "Union internationale de chimie pure et appliquée" et des CA "Chemical Abstracts").
Indiquer la dénomination chimique précisée à l'annexe I de la directive 65/548/CEE ou, si la dénomination ne figure pas dans cette directive, conformément à la nomenclature de l'UICPA et des CA.
1.5. Numéro(s) de code développement du fabricant.
Indiquer les numéros de code utilisés pour identifier la substance active et les préparations éventuellement disponibles contenant la substance active, pendant le travail de développement. Préciser pour chaque numéro de code indiqué le matériel auquel il se réfère, la période pendant laquelle il a été utilisé et les Etats membres ou autres pays dans lesquels il a été ou est encore utilisé.
1.6. Numéro CAS, numéro CEE et numéro CIMAC (si disponibles).
Indiquer le numéro CAS, le numéro CEE (Einecs ou Elincs) et le numéro CIMAC, lorsqu'ils existent.
1.7. Formule moléculaire et formule développée, masse moléculaire.
Indiquer la formule moléculaire, la masse moléculaire et la formule développée de la substance active et, le cas échéant, la formule développée de chaque stéréo-isomère et isomère optique présent dans la substance active.
1.8. Méthode de fabrication de la substance active (procédé de synthèse).
Indiquer pour chaque installation la méthode de fabrication, en termes d'identité des matières de départ, de procédés chimiques utilisés ainsi que d'identité des sous-produits et des impuretés présents dans le produit fini. L'information sur l'ingénierie des procédés n'est généralement pas requise.
Lorsque l'information fournie concerne un système de production pilote, l'information requise doit de nouveau être fournie lorsque les méthodes et procédures de production à l'échelle industrielle se sont stabilisées.
1.9. Spécification de la pureté de la substance active exprimée en grammes par kilogramme.
Indiquer la teneur minimale, en g/kg de substance active pure (à l'exclusion des isomères inactifs), de la matière manufacturée entrant dans la fabrication des produits préparés.
Lorsque l'information fournie concerne un système de production pilote, l'information requise doit de nouveau être fournie à la Commission et aux Etats membres lorsque les méthodes et procédures de production à l'échelle industrielle se sont stabilisées et si les changements intervenus dans la production modifient la spécification de la pureté.
1.10. Identité des isomères, impuretés et additifs (par exemple des stabilisants), avec la formule développée et la teneur exprimée en grammes par kilogramme.
Indiquer la teneur maximale en g/kg des isomères inactifs ainsi que le ratio entre la teneur en isomères/diastéréo-isomères, le cas échéant. En outre, indiquer la teneur maximale en g/kg de chaque composant autre que les additifs, y compris les sous-produits et les impuretés. Pour les additifs, indiquer la teneur en g/kg.
Pour chaque composant présent à raison de 1 g/kg ou plus, fournir les informations suivantes, le cas échéant :
- dénomination chimique conformément à la nomenclature de l'UICPA et des CA ;
- nom commun ISO ou nom commun proposé, s'il est disponible ;
- numéro CAS, numéro CEE (Einecs ou Elincs) et numéro CIMAC, s'ils sont disponibles ;
- formule moléculaire et formule développée ;
- masse moléculaire ;
- teneur maximale en g/kg.
Lorsque le procédé de fabrication est tel que des impuretés et des sous-produits particulièrement indésirables en raison de leurs propriétés toxicologiques, écotoxicologiques ou environnementales peuvent être présents dans la substance active, déterminer et indiquer la teneur en chacun de ces composés. Dans ces cas, indiquer les méthodes d'analyse utilisées et les limites de détermination, qui doivent être suffisamment faibles pour chaque composé important. De plus, fournir les informations suivantes, le cas échéant :
- dénomination chimique conformément à la nomenclature de l'UICPA et des CA ;
- nom commun ISO ou nom commun proposé, s'il est disponible ;
- numéro CAS, numéro CEE (Einecs ou Elincs) et numéro CIMAC, s'ils sont disponibles ;
- formule moléculaire et formule développée ;
- masse moléculaire ;
- teneur maximale en g/kg.
Lorsque l'information fournie concerne un système de production pilote, les informations requises doivent de nouveau être fournies lorsque les méthodes et procédures de production à l'échelle industrielle se sont stabilisées, si les changements intervenus dans la production modifient la spécification de la pureté.
Lorsque les informations fournies ne permettent pas d'identifier pleinement un composant, par exemple des condensats, fournir des informations détaillées sur la composition de chacun de ces composants.
Lorsque des composants sont ajoutés à la substance active, avant la fabrication du produit préparé, pour protéger sa stabilité et faciliter sa manipulation, il y a lieu d'indiquer également leur dénomination commerciale. De plus, fournir les informations suivantes sur ces additifs, le cas échéant :
- dénomination chimique conformément à la nomenclature de l'UICPA et des CA ;
- nom commun ISO ou nom commun proposé, s'il est disponible ;
- numéro CAS, numéro CEE (Einecs ou Elincs) et numéro CIMAC, s'ils sont disponibles ;
- formule moléculaire et formule développée ;
- masse moléculaire ;
- teneur maximale en g/kg.
Pour les composants ajoutés, autres que la substance active et que les impuretés résultant du procédé de fabrication, indiquer la fonction du composant (additif) :
- agent antimoussant ;
- antigel ;
- liant ;
- tampon ;
- agent dispersant ;
- stabilisant ;
- autres (préciser).
1.11. Profil analytique des lots.
Analyser des échantillons représentatifs de la substance active pour déterminer leur teneur en substance active pure, isomères inactifs, impuretés et additifs, selon le cas. Les résultats d'analyse transmis doivent comprendre les données quantitatives, en termes de teneur en g/kg, pour tous les composants présents à raison de plus de 1 g/kg ; normalement, ils doivent porter sur 98 p. 100 au moins de la matière analysée. Déterminer et communiquer la teneur réelle en composants particulièrement indésirables en raison de leurs propriétés toxicologiques, écotoxicologiques ou environnementales. Les données communiquées doivent comprendre les résultats d'analyse d'échantillons individuels ainsi qu'un sommaire de ces données, destiné à indiquer la teneur minimale ou maximale et typique en chaque composant important, selon le cas.
Lorsqu'une substance active est produite dans plusieurs installations, il convient de fournir ces informations séparément pour chacune des installations.
Par ailleurs, si nécessaire et possible, il convient d'analyser des échantillons de la substance active produite en laboratoire ou dans des systèmes pilotes de production lorsque ces matériels ont servi à fournir des données toxicologiques ou écotoxicologiques.
2. Propriétés physiques et chimiques de la substance active.
i) L'information fournie doit décrire les propriétés physiques et chimiques des substances actives ; avec d'autres informations importantes, elle doit permettre de les caractériser. En particulier, l'information fournie doit permettre :
- d'identifier les risques physiques, chimiques et techniques liés aux substances actives ;
- de classer les substances actives sur le plan du risque ;
- de choisir les restrictions et conditions appropriées à associer à l'insertion de substances ;
- de spécifier les phrases appropriées sur le plan du risque et de la sécurité.
Les informations et données visées sont requises pour toutes les substances actives, sauf précision contraire.
ii) Les informations fournies, associées à celles concernant les préparations importantes, doivent permettre d'identifier les risques physiques, chimiques et techniques liés aux préparations, de classer ces dernières et d'établir que des préparations peuvent être utilisées sans difficulté inutile et sont telles que l'homme, les animaux et l'environnement soient exposés le moins possible, compte tenu du mode d'utilisation.
iii) Indiquer dans quelle mesure les substances actives dont l'insertion est demandée sont conformes aux spécifications correspondantes de la FAO (Organisation des Nations unies pour l'alimentation et l'agriculture). Préciser et justifier les divergences par rapport à ces spécifications.
iv) Dans des cas précis, les tests doivent être réalisés sur une substance active purifiée répondant à des spécifications données. Dans ces cas, il y a lieu d'indiquer les principes de la (des) méthode(s) de purification. Indiquer le degré de pureté de cette matière d'essai, qui doit être aussi élevé que le permet la meilleure technologie disponible. Fournir une justification en bonne et due forme dans les cas où le degré de pureté est inférieur à 980 g/kg.
Cette justification doit démontrer que toutes les possibilités techniquement réalisables et acceptables de production de la substance active pure ont été envisagées.
2.1. Point de fusion et point d'ébullition.
2.1.1. Déterminer et indiquer le point de fusion ou, le cas échéant, le point de congélation ou de solidification de la substance active purifiée, conformément à la méthode CEE A1. Les mesures doivent être effectuées jusqu'à 360° C.
2.1.2. Pour les substances actives qui sont liquides, déterminer et indiquer, le cas échéant, le point d'ébullition de ces substances conformément à la méthode CEE A 2. Les mesures doivent être effectuées jusqu'à 360° C.
2.1.3. Lorsque le point de fusion et/ou le point d'ébullition ne peuvent être déterminés pour des raisons de décomposition ou de sublimation, indiquer la température à laquelle se produit la décomposition ou la sublimation.
2.2. Densité relative.
Pour les substances actives liquides ou solides, déterminer et indiquer la densité relative de la substance active purifiée conformément à la méthode CEE A 3.
2.3. Pression de vapeur (en Pa) volatilité (par exemple constante de la loi de Henry).
2.3.1. Indiquer la pression de vapeur de la substance active purifiée, selon la méthode CEE A 4. Lorsque cette pression est inférieure à 10-5 Pa, la pression de vapeur à 20 ou 25 °C peut être estimée par une courbe de pression de vapeur.
2.3.2. Pour les substances actives solides ou liquides, déterminer la volatilité (constante de la loi de Henry) de la substance active purifiée, ou la calculer à partir de sa solubilité dans l'eau et de la pression de vapeur et l'indiquer (en Pa x m3 x mol-1).
2.4. Aspect (état physique, couleur et odeur, s'ils sont connus).
2.4.1. Donner une description de la couleur, le cas échéant, et de l'état physique de la substance active manufacturée et de la substance active purifiée.
2.4.2. Donner une description de toute odeur associée à la substance active manufacturée et à la substance active purifiée, constatée lors de la manipulation des matières en laboratoire ou dans les installations de production.
2.5. Spectres ultraviolet-visible (UV-VIS), infrarouge (IR), résonance magnétique nucléaire (R.M.N.), spectrométrie de masse (S.M.), extinction moléculaire aux longueurs d'onde adéquates.
2.5.1. Déterminer et indiquer les spectres suivants, avec un tableau des caractéristiques du signal nécessaires à l'interprétation : ultraviolet-visible (UV-VIS), infrarouge (IR), résonance magnétique nucléaire (R.M.N.) et spectrométrie de masse (S.M.) de la substance active purifiée et extinction moléculaire aux longueurs d'onde adéquates. Déterminer et indiquer les longueurs d'onde auxquelles l'extinction moléculaire a lieu dans le spectre ultraviolet-visible ; si nécessaire, y inclure une longueur d'onde à la plus haute valeur d'absorption au-dessus de 290 nm.
Pour les substances actives qui sont des isomères optiques résolus, mesurer et indiquer leur pureté optique.
2.5.2. Déterminer et indiquer les spectres d'absorption UV-VIS, IR R.M.N. et S.M. s'ils sont nécessaires pour l'identification de toutes les impuretés considérées comme importantes sur le plan toxicologique, écotoxicologique ou environnemental.
2.6. Solubilité dans l'eau, notamment influence du pH (4 à 10) sur la solubilité.
Déterminer et indiquer, conformément à la méthode CEE A 6, la solubilité dans l'eau des substances actives purifiées à la pression atmosphérique. Effectuer ces déterminations dans la gamme neutre (c'est-à-dire dans de l'eau distillée en équilibre avec le dioxyde de carbone atmosphérique). Lorsque la substance active est capable de former des ions, effectuer les déterminations dans la gamme acide (pH 4 à 6) et dans la gamme alcaline (pH 8 à 10).
Lorsque la stabilité de la substance active dans les milieux aqueux ne permet pas de déterminer la solubilité dans l'eau, fournir une justification reposant sur les données d'essai.
2.7. Solubilité dans les solvants organiques.
Déterminer et indiquer la solubilité des substances actives fabriquées dans les solvants organiques suivants, à une température de 5 à 25° C, si elle est inférieure à 250 g/kg ; préciser la température appliquée :
- hydrocarbure aliphatique : de préférence n-heptane ;
- hydrocarbure aromatique : de préférence xylène ;
- hydrocarbure halogène : de préférence 1.2-dichloro-éthane ;
- alcool : de préférence méthanol ou alcool isopropylique ;
- cétone : de préférence acétone ;
- ester : de préférence acétate d'éthyle.
Si un ou plusieurs de ces solvants ne convient pas à une substance active donnée (par exemple s'il réagit avec la substance testée), il(s) peut (peuvent) être remplacé(s) par d'autres solvants. Dans ce cas, justifier les choix effectués au niveau de la structure et de la polarité des solvants.
2.8. Coefficient de partage n-octanol/eau, notamment influence du pH (4 à 10).
Déterminer le coefficient de partage n-octanol/eau de la substance active purifiée et l'indiquer selon la méthode CEE A 8. Analyser l'incidence du pH (4 à 10) lorsque la substance est acide ou basique selon sa valeur pKa (12 pour les acides, 2 pour les bases).
2.9. Stabilité dans l'eau, taux d'hydrolyse, dégradation photochimique, proportion et identité du (des) produits(s) de dégradation, constante de dissociation, notamment influence du pH (4 à 9).
2.9.1. Déterminer le taux d'hydrolyse des substances actives purifiées (généralement substance active marquée, d'une pureté 95 p. 100, pour chacune des valeurs du pH 4, 7 et 9, en atmosphère stérile et en l'absence de lumière, et l'indiquer conformément à la méthode CEE C 7. Pour les substances ayant un faible taux d'hydrolyse, ce taux peut être déterminé à 50° C ou à une autre température appropriée.
Si une dégradation se produit à 50 °C, déterminer le taux de dégradation à une autre température et tracer un graphique d'Arrhenius pour permettre d'estimer l'hydrolyse à 20° C. Indiquer l'identité des produits formés par hydrolyse et la constante de vitesse observée. Indiquer aussi la valeur DT50 estimée.
2.9.2. Pour les composés ayant un coefficient d'absorption moléculaire (décimal) (E) 10 (1 x mol-1 x cm-1) à une longueur d'onde 290 nm, déterminer et indiquer la phototransformation directe dans l'eau purifiée (par exemple distillée), à une température comprise entre 20 et 25° C, d'une substance active purifiée, généralement marquée à la lumière artificielle et en atmosphère stérile, si nécessaire en utilisant un agent de solubilisation. Ne pas utiliser d'activateurs tels que l'acétone comme co-solvant ou comme agent de solubilisation. La source de lumière doit simuler la lumière du soleil et être équipée de filtres qui excluent les radiations à des longueurs d'ondes 290 nm. Indiquer l'identité des produits de dégradation formés qui sont présents à tout moment pendant la réalisation de l'étude dans des quantités 10 p. 100 de la substance active ajoutée, fournir un bilan matière permettant de tenir compte de 90 p. 100 au moins de la radio-activité appliquée et indiquer la demi-vie photochimique.
2.9.3. Si cela est nécessaire pour étudier la phototransformation directe, déterminer et indiquer le rendement quantique de la photodégradation directe dans l'eau, les calculs permettant d'estimer la durée de vie théorique de la substance active dans la couche supérieure des systèmes aqueux et la durée de vie réelle de la substance.
La méthode est décrite dans les directives modifiées de la FAO relatives aux critères écotoxicologiques d'homologation des pesticides.
2.9.4. Lorsqu'une dissociation dans l'eau se produit, déterminer et indiquer conformément à la ligne directrice n° 112 de l'Organisation de coopération et de développement économiques (O.C.D.E.) la (les) constante(s) de dissociation (valeurs pKa) des substances actives purifiées. Indiquer l'identité des produits de dissociation formés, reposant sur des considérations théoriques. Si la substance active est un sel, indiquer la valeur pKa du principe actif.
2.10. Stabilité dans l'air, dégradation photochimique, identité du (des) produit(s) de dégradation.
Présenter une estimation de la dégradation photochimique oxydative (autotransformation indirecte) de la substance active.
2.11. Inflammabilité, y compris auto-inflammabilité.
2.11.1. Déterminer l'inflammabilité des substances actives fabriquées qui sont solides, gazeuses ou qui dégagent des gaz très inflammables, et l'indiquer conformément à la méthode CEE A 10, A 11 ou A 12, selon le cas.
2.11.2. Déterminer l'auto-inflammabilité des substances actives manufacturées et l'indiquer conformément à la méthode CEE A 15 ou A 16, selon le cas et/ou, si nécessaire, selon l'essai en cage Bowes-Cameron des Nations unies (recommandations des Nations unies sur le transport des marchandises dangereuses, chapitre 14, n° 14.3.4).
2.12. Point d'éclair.
Déterminer le point d'éclair des substances actives manufacturées ayant un point de fusion inférieur à 40° C et l'indiquer conformément à la méthode CEE A 9 ; il convient de n'utiliser que des méthodes en vase clos.
2.13. Propriétés explosives.
Si nécessaire, déterminer et indiquer conformément à la méthode CEE A 14 les propriétés explosives des substances actives manufacturées.
2.14. Tension superficielle.
Déterminer et indiquer la tension superficielle selon la méthode CEE A 5.
2.15. Propriétés oxydantes.
Déterminer les propriétés oxydantes des substances actives manufacturées et les indiquer conformément à la méthode CEE A 17, sauf lorsque l'examen de leur formule développée établit de manière relativement incontestable que la substance active considérée est incapable de réagir exothermiquement avec une matière combustible. Dans ces cas, il suffit de fournir ces informations pour justifier la non-détermination des propriétés oxydantes de la substance.
(Arrêté du 1er février 1996, article 1er)
i) L'information fournie doit indiquer à quelles fins il est envisagé d'utiliser les préparations contenant la substance active ou à quelles fins elles vont l'être, et préciser quels seront la dose appliquée et le mode d'utilisation prévus ou proposés.
ii) L'information fournie doit préciser les méthodes et précautions normales à suivre dans la manipulation, le stockage et le transport de la substance active.
iii) Les études, données et informations présentées ainsi que d'autres études, données et informations pertinentes doivent préciser et justifier les méthodes et précautions à suivre en cas d'incendie et identifier les produits de combustion alors obtenus. Il convient de prévoir, en fonction de la structure chimique et des propriétés physiques et chimiques de la substance active, les produits de combustion susceptibles de se former en cas d'incendie.
iv) Les études, données et informations présentées ainsi que d'autres études, données et informations pertinentes doivent démontrer que les mesures proposées conviennent dans des situations d'urgence.
v) Les informations et données précitées sont requises pour toutes les substances actives, sauf indication contraire.
3.1. Fonction, par exemple fongicide, herbicide, insecticide, répulsif, régulateur de croissance.
La fonction, choisie parmi celles énumérées ci-après, doit être précisée :
- acaricide ;
- bactéricide ;
- fongicide ;
- herbicide ;
- insecticide ;
- molluscicide ;
- nématicide ;
- régulateur de croissance végétale ;
- répulsif ;
- rodenticide ;
- médiateur chimique ;
- taupicide ;
- virucide ;
- autres (à préciser).
3.2. Effets sur les organismes nuisibles, par exemple poison par contact, par inhalation, poison stomacal, fongitoxique ou fongistatique, etc., systémique ou non chez les végétaux.
3.2.1. Indiquer la nature des effets sur les organismes nuisibles :
- action par contact ;
- action par ingestion ;
- action par inhalation ;
- action fongitoxique ;
- action fongistatique ;
- déshydratant ;
- inhibiteur de la reproduction ;
- autres (à préciser).
3.2.2. Indiquer si la substance active est transportée dans des végétaux et, le cas échéant, si ce déplacement est apoplastique, symplastique ou les deux.
3.3. Domaine d'utilisation envisagé, par exemple champ, serre, stockage de produits végétaux, jardinage.
Préciser, parmi ceux indiqués ci-après, le(s) domaine(s) d'utilisation, actuel(s) et proposé(s), des préparations contenant la substance active :
- utilisation en pleine terre, comme en agriculture, horticulture, sylviculture, viticulture ;
- serre ;
- agrément ;
- désherbage des terres non cultivées ;
- jardinage ;
- plantes d'intérieur ;
- stockage de produits végétaux ;
- autres (à préciser).
3.4. Organismes nuisibles combattus et cultures et produits protégés ou traités.
3.4.1. Préciser l'utilisation actuelle et envisagée en termes de cultures, groupes de cultures, végétaux ou produits végétaux traités et, le cas échéant, protégés.
3.4.2. Le cas échéant, spécifier les organismes nuisibles contre lesquels une protection est assurée.
3.4.3. Le cas échéant, indiquer les effets obtenus, par exemple la suppression des pousses, le retardement de la maturation, la diminution de la longueur des tiges, une meilleure fécondation, etc.
3.5. Mode d'action.
3.5.1. Dans la mesure où il a été élucidé, indiquer le mode d'action de la substance active au niveau, le cas échéant, du (des) mécanisme(s) biochimique(s) et physiologique(s) ainsi que du (des) procédé(s) biochimique(s). S'ils sont disponibles, indiquer les résultats des études expérimentales en la matière.
3.5.2. Lorsqu'on sait que pour exercer l'effet recherché, la substance active doit être transformée en métabolite ou en produit de dégradation après application ou utilisation des préparations qui la contiennent, fournir au sujet du métabolite ou produit de dégradation actif les informations suivantes, faisant référence et appel aux informations contenues aux points 5.6, 5.11, 6.1, 6.2, 6.7, 7.1, 7.2 et 9, le cas échéant :
- dénomination chimique conformément à la nomenclature de l'UICPA et du CA ;
- nom commun ISO ou nom commun proposé ;
- numéro CAS, numéro CEE (Einecs et Elincs) et numéro CIMAC, s'il est disponible ;
- formule empirique et formule développée ;
- masse moléculaire.
3.5.3. Fournir les informations disponibles sur la formation des métabolites et produits de dégradation actifs et notamment :
- les procédés, mécanismes et réactions impliqués ;
- les données cinétiques et autres données concernant la vitesse de conversion et, s'il est connu, le facteur limitant pour la vitesse ;
- les facteurs environnementaux et ceux ayant une incidence sur la vitesse et l'importance de la conversion.
3.6. Informations sur l'apparition ou l'apparition éventuelle du développement d'une résistance et stratégies de réponse.
Lorsqu'il en existe, fournir des informations sur l'apparition éventuelle du développement d'une résistance ou d'une résistance croisée.
3.7. Méthodes et précautions recommandées en matière de manipulation, stockage, transport ou incendie.
Fournir une fiche de données de sécurité visée à l'article 27 de la directive 67/548/CEE du Conseil pour toutes les substances actives.
3.8. Procédures de destruction ou de décontamination de la substance active.
3.8.1. Incinération contrôlée :
Dans de nombreux cas, la manière préférée ou l'unique manière d'éliminer en toute sécurité des substances actives, des matières contaminées ou des emballages contaminés est de les soumettre à une incinération contrôlée dans un incinérateur agréé.
Lorsque la teneur en halogènes de la substance active est supérieure à 60 p. 100, indiquer le comportement pyrolytique de la substance active dans des conditions contrôlées (y compris, le cas échéant, l'apport précis en oxygène et le temps de séjour fixe) à 800 °C et la teneur en dibenzo-p-dioxines et dibenzo-furanes polyhalogénés dans les produits de la pyrolyse. Le demandeur doit fournir des instructions détaillées sur la sécurité d'élimination.
3.8.2. Divers :
Décrite en détail les autres méthodes d'élimination de la substance active, d'emballages contaminés et de matières contaminées, s'il en est proposé. Fournir des données sur ces méthodes permettant d'établir leur efficacité et leur sécurité.
3.9. Mesures d'urgence en cas d'accident.
Indiquer les procédures de décontamination de l'eau, en cas d'accident.
4. Méthodes d'analyse
(Arrêté du 1er février 1996, article 1er et Arrêté du 12 janvier 1998, article 1er I)
Introduction.
Les dispositions du présent point s'appliquent exclusivement aux méthodes d'analyse requises pour le contrôle et le suivi postérieurs à l'autorisation.
Pour les méthodes d'analyse utilisées pour la production des données requises par la présente directive ou à d'autres fins, le demandeur est tenu de fournir une justification de la méthode utilisée ; si nécessaire, des directives spécifiques seront mises au point pour de telles méthodes sur la base des mêmes normes que celles requises pour les méthodes de contrôle et de suivi postérieurs à l'autorisation.
La description des méthodes d'analyse doit être fournie et contenir toutes les données utiles concernant l'équipement, les matériels et les conditions d'application.
Ces méthodes doivent, autant que possible, suivre l'approche la plus simple, être peu onéreuses et faire appel à des équipements courants.
Les définitions suivantes s'appliquent aux fins du présent chapitre :
Impuretés : tout composant autre que la substance active pure, comprise dans la substance active technique (y compris les isomères non actifs) provenant du processus de fabrication ou de la dégradation survenue durant le stockage ;
Impuretés caractéristiques : impuretés posant des problèmes d'ordre toxicologique et/ou écotoxicologique ou environnemental ;
Impuretés significatives : impuretés représentant une quantité dans la substance active technique égale ou supérieure à 1 g/kg ;
Métabolites : métabolites, y compris les produits résultant de la dégradation ou de la réaction de la substance active ;
Métabolites caractéristiques : métabolites posant des problèmes d'ordre toxicologique et/ou écotoxicologique ou environnemental.
A la demande, les échantillons suivants doivent être fournis :
i) Des étalons pour analyse de la substance active pure ;
ii) Des échantillons de la substance active technique ;
iii) Des étalons pour l'analyse des métabolites caractéristiques et de tous les autres composants compris dans la définition de résidu ;
iv) Si disponibles, des échantillons des substances de référence des impuretés caractéristiques.
4.1. Méthodes d'analyse de la substance active technique.
Les définitions ci-après sont applicables à la présente section :
i) Spécificité.
La spécificité est la capacité de la méthode de discerner la substance recherchée à mesurer des autres substances.
ii) Linéarité.
La linéarité est la capacité de la méthode, dans une plage donnée, de fournir une corrélation linéaire acceptable entre les résultats et la concentration d'analyte dans l'échantillon.
iii) Exactitude.
L'exactitude est la mesure dans laquelle la valeur obtenue par l'analyte dans un échantillon correspond à la valeur de référence reconnue (cf. par ex. ISO 5725).
iv) Précision.
La précision est le degré de concordance des résultats de tests indépendants pratiqués dans les conditions prescrites.
Répétabilité : la précision obtenue dans des conditions répétables, c'est-à-dire des conditions dans lesquelles les résultats de tests indépendants sont obtenus par l'application de la même méthode à un matériel d'essai identique, dans un même laboratoire et par un même opérateur utilisant un même équipement à de brefs intervalles de temps.
La reproductibilité n'est pas requise pour la substance active technique (pour la définition de la reproductibilité, voir ISO 5725).
4.1.1. Il y a lieu de présenter, et de décrire dans leur intégralité, les méthodes qui permettent de déterminer la substance active pure présente dans la substance active technique conformément au dossier présenté aux fins d'inclusion de la substance à l'annexe I de la directive 91/414/CEE. L'applicabilité des méthodes actuelles de la CIMAP doit être signalée.
4.1.2. Il convient également de présenter des méthodes qui permettent de doser dans la substance active technique les impuretés et les additifs (les stabilisants, par exemple) significatifs et/ou caractéristiques.
4.1.3. Spécificité, linéarité, exactitude et répétabilité.
4.1.3.1. La spécificité des méthodes présentées doit être démontrée et consignée. Il y a lieu, en outre, de déterminer l'ampleur de l'interférence des autres substances (isomètres, impuretés, additifs) présentes dans la substance active technique.
Les interférences d'autres composantes peuvent être considérées comme des erreurs systématiques dans l'évaluation de l'exactitude des méthodes proposées pour le dosage de la substance active pure dans la substance active technique ; néanmoins, une explication doit être donnée pour toute interférence contribuant pour plus de 3 % de la quantité totale dosée.
Le degré de l'interférence doit être déterminé également pour les méthodes de détermination des impuretés.
4.1.3.2. La linéarité des méthodes proposées dans une plage appropriée doit être déterminée et consignée. Pour le dosage de la substance pure, la plage d'étalonnage doit dépasser (d'au moins 20 %) la teneur nominale la plus élevée et la plus basse de la substance recherchée dans les solutions à analyser en cause. Pour l'étalonnage, ont doit effectuer une double mesure d'au moins trois concentrations différentes ou une mesure simple de cinq concentrations. Les procès-verbaux doivent contenir l'équation de la courbe d'étalonnage et le coefficient de corrélation ainsi que des documents relatifs à l'analyse, représentatifs et dûment étiquetés, par exemple des chromatogrammes.
4.1.3.3. Le critère d'exactitude est requis pour le dosage de la substance pure et les impuretés significatives et/ou importantes dans la substance active technique.
4.1.3.4. Au moins cinq dosages sont normalement requis pour la répétabilité du dosage de la substance active pure. L'écart type relatif (% ETR) doit être mentionné. Les valeurs aberrantes observées par une méthode appropriée (le test de Dixons ou de Grubbs, par exemple) peuvent être négligées, mais leur écart doit toujours être signalé et leur apparition doit faire l'objet d'une tentative d'explication.
4.2. Méthodes de détection des résidus.
Ces méthodes doivent permettre de détecter la substance active et/ou les métabolites caractéristiques. Pour chaque méthode et pour chaque matière représentative, il y a lieu de déterminer expérimentalement et de consigner la spécificité, la précision, la possibilité de récupération et la limite de détermination.
En principe, les méthodes proposées doivent permettre la détection de multiples résidus ; une méthode multirésidus standard doit faire l'objet d'une évaluation et d'un rapport pour voir si elle convient. Lorsque ces méthodes ne sont pas des méthodes multirésidus ou sont incompatibles avec celles-ci, une méthode de remplacement doit être proposée. Si cette exigence aboutit à la proposition d'un nombre de méthodes excessif pour des composés particuliers, une "méthode relative à la partie commune" peut être acceptable.
Les définitions ci-après sont applicables au présent chapitre.
i) Spécificité.
La spécificité est la capacité d'une méthode de discerner la substance recherchée à mesurer des autres substances.
ii) Précision.
La précision est le degré de concordance des résultats de tests indépendants obtenus dans des conditions déterminées.
La répétabilité est la précision obtenue dans des conditions répétables, c'est-à-dire des conditions dans lesquelles les résultats de tests indépendants sont obtenus par l'application d'une même méthode à un matériel d'essai identique, dans un même laboratoire et par un même opérateur utilisant un même équipement à de brefs intervalles de temps.
Reproductibilité : étant donné que la définition de la reproductibilité dans les publications pertinentes (par exemple dans ISO 5725) n'est généralement pas praticable pour des méthodes d'analyse de résidu, la reproductibilité dans le contexte de la présente directive se définit comme une validation de la répétabilité de la récupération de matières représentatives et à des niveaux de concentration représentatifs par au moins un laboratoire qui est indépendant de celui qui a initialement validé l'étude (ce laboratoire indépendant peut être dans la même firme) (validation de laboratoires indépendants).
iii) Récupération.
Le pourcentage de la quantité de substance active ou de métabolite caractéristique ajouté initialement à un échantillon de la matrice appropriée, qui ne contient aucun niveau détectable de la substance recherchée.
iv) Limite de détermination.
La limite de détermination (souvent appelée limite de quantification) est la plus faible concentration testée à laquelle on obtient une récupération moyenne acceptable (normalement 70 à 110 % avec un écart type relatif de préférence 20 % ; dans certains cas justifiés, des taux moyens de récupération inférieurs ou supérieurs ainsi que des écarts types relatifs supérieurs peuvent être acceptables).
4.2.1. Résidus dans et/ou sur des végétaux, produits végétaux, denrées alimentaires (d'origine végétale et animale), aliments pour animaux :
Les méthodes proposées doivent convenir pour le dosage de tous les composants compris dans la définition du résidu proposée conformément aux dispositions du chapitre 6, points 6.1 et 6.2, en vue de permettre aux Etats membres de déterminer la conformité avec les limites maximales de résidus établies ou de déterminer le niveau de transfert aux travailleurs.
La spécificité des méthodes doit permettre le dosage de tous les composants compris dans la définition du résidu et/ou des métabolites pertinents ; une technique supplémentaire de confirmation doit être appliquée, si appropriée.
La répétabilité doit être déterminée et mentionnée. Les échantillons d'essai d'analyse identiques peuvent, autant que possible, être préparés à partir d'un même échantillon traité sur le terrain contenant les résidus rencontrés. Par ailleurs, les échantillons d'essai peuvent être préparés à partir d'un échantillon commun non traité dont les aliquotes ont été portées au(x) niveau(x) requis.
Les résultats d'une validation de laboratoire indépendant doivent être mentionnés.
La limite de détermination ainsi que la récupération individuelle et moyenne doivent être déterminées et consignées. L'écart type relatif global et l'écart type relatif pour chaque niveau de supplémentation doivent être mentionnés.
4.2.2. Résidus présents dans le sol :
Il y a lieu de proposer des méthodes d'analyse du sol permettant de déterminer le précurseur et/ou les métabolites importants ;
La spécificité des méthodes doit permettre la détermination du précurseur et/ou métabolites importants à l'aide d'une technique de confirmation supplémentaire, si appropriés ;
La répétabilité, la récupération et la limite de détermination, y compris la récupération individuelle et la récupération moyenne, doivent être déterminées et mentionnées. L'écart type relatif global ainsi que l'écart type relatif pour chaque niveau de supplémentation doivent être déterminés expérimentalement et consignés ;
La limite de détermination proposée ne doit pas dépasser une concentration qui a un impact inacceptable pour les organismes non ciblés ou à cause des effets phytotoxiques. Normalement la limite de détermination proposée ne devrait pas dépasser 0,05 mg/kg.
4.2.3. Résidus présents dans l'eau (y compris l'eau potable, l'eau souterraine et l'eau de surface) :
Il y a lieu de proposer des méthodes d'analyse de l'eau permettant de déterminer le précurseur et/ou les métabolites caractéristiques ;
La spécificité des méthodes doit permettre la détermination du précurseur et/ou métabolites importants à l'aide d'une technique de confirmation supplémentaire, si approprié ;
La répétabilité, la récupération et la limite de détermination, y compris la récupération individuelle et la récupération moyenne, doivent être déterminées et mentionnées. L'écart type relatif global ainsi que l'écart type relatif doivent être déterminés expérimentalement et mentionnés pour chaque degré de supplémentation ;
Pour l'eau potable, la limite de détermination proposée ne doit pas dépasser la concentration maximale la moins élevée mentionnée dans le point 2.5.1.2, partie C, des principes uniformes. Pour l'eau de surface, elle ne doit pas dépasser une concentration mentionnée au point 2.5.1.3, partie C, des principes uniformes.
4.2.4. Résidus présents dans l'air :
Il y a lieu de proposer des méthodes de détermination de la substance active et/ou des métabolites caractéristiques présentes dans l'air pendant ou peu de temps après l'application sauf si on peut justifier que l'exposition des opérateurs ou spectateurs est peu probable ;
La spécificité des méthodes doit permettre la détermination du précurseur et/ou métabolites caractéristiques à l'aide d'une technique de confirmation supplémentaire si approprié ;
La répétabilité, la récupération et la limite de détermination, y compris la récupération individuelle et la récupération moyenne, doivent être déterminées et mentionnées. L'écart type relatif global ainsi que l'écart type relatif doivent être déterminés expérimentalement et mentionnés pour chaque degré de supplémentation ;
La limite de détermination proposée doit tenir compte de valeurs limites pertinentes pour la santé ou de degrés d'exposition caractéristiques.
4.2.5. Résidus présents dans les liquides et tissus organiques :
Il y a lieu de proposer des méthodes d'analyse appropriées lorsqu'une substance active est classée comme toxique ou hautement toxique ;
La spécificité des méthodes doit permettre la détermination du précurseur et/ou des métabolites importants à l'aide d'une technique de confirmation, si approprié ;
La répétabilité, la récupération et la limite de détermination, y compris la récupération individuelle et la récupération moyenne, doivent être déterminées et mentionnées. L'écart type relatif global ainsi que l'écart type relatif doivent être déterminés expérimentalement et consignés pour chaque degré de supplémentation.
Article Annexe I
Modifié par
5. Etudes toxicologiques et de métabolisme
(Arrêté du 1er février 1996, article 1er)
Introduction.
i) Les informations fournies jointes à celles concernant une ou plusieurs préparations contenant la substance active doivent être suffisantes pour permettre une évaluation des risques pour l'homme, découlant de la manipulation et de l'utilisation de produits phytopharmaceutiques contenant la substance active, et du risque pour l'homme dû aux résidus contenus dans les aliments et dans l'eau. En outre, les informations doivent être suffisantes pour :
- permettre une décision quant à l'inclusion éventuelle de la substance active dans la liste communautaire des substances actives ;
- fixer les conditions ou restrictions appropriées liées à toute inclusion dans la liste communautaire des substances actives ;
- classer la substance active quant au danger ;
- fixer une dose journalière acceptable pertinente (DJA) pour l'homme ;
- fixer des niveaux acceptables d'exposition de l'opérateur (NAEO) ;
- fixer les symboles des dangers, les indications relatives aux dangers et les phrases types relatives à la nature des risques et aux conseils de prudence pour la protection de l'homme, des animaux et de l'environnement à faire figurer sur l'emballage (conteneurs) ;
- définir les mesures adéquates en soins d'urgence ainsi que les mesures appropriées concernant le diagnostic correct et les traitements thérapeutiques en cas d'empoisonnement chez l'homme.
- permettre une évaluation concernant la nature et l'ampleur des risques pour l'homme, les animaux (espèces normalement nourries et élevées ou consommées par l'homme) et des risques pour d'autres espèces de vertébrés non ciblés.
ii) Il est nécessaire d'examiner et de relater tous les effets néfastes possibles découverts au cours des études toxicologiques de routine (y compris les effets sur les organes et certains systèmes déterminés, tels que l'immunotoxicité et la neurotoxicité) et d'entreprendre et de relater les études supplémentaires qui peuvent être nécessaires pour analyser le mécanisme probable en cause, de fixer des NOAEL (niveaux sans effet néfaste observable), et d'estimer l'importance de ces effets. Toutes les données biologiques et informations disponibles pertinentes pour l'évaluation du profil toxicologique de la substance testée doivent être relatées.
iii) Compte tenu de l'influence que peuvent avoir les impuretés sur le comportement toxicologique, il est essentiel que pour toute étude proposée soit fournie une description détaillée (spécification) du matériel utilisé, mentionné à la section 1, point 11. Les essais doivent être effectués avec la substance active de ladite spécification, qui sera utilisée pour la fabrication des préparations à autoriser, sauf si une substance marquée radioactivement est exigée ou autorisée.
iv) Si des études sont effectuées avec une substance active produite dans le laboratoire ou dans un système de production d'usine pilote, les études doivent être répétées avec la substance active telle qu'elle est fabriquée, sauf s'il peut être prouvé que la substance d'essai utilisée est fondamentalement la même, aux fins d'essai et d'évaluation de la toxicité. En cas d'incertitude, des études appropriées permettant de faire la liaison doivent être présentées pour permettre d'arrêter une décision quant à l'éventuelle nécessité de répéter les études.
v) Dans le cas d'études dans lesquelles le dosage s'étend sur une période, le dosage doit être fait de préférence avec un seul lot de substance active, si la stabilité le permet.
vi) Pour toutes les études, la dose réelle employée, exprimée en milligrammes par kilogramme de poids corporel, ainsi que dans d'autres unités appropriées, doit être relatée. Si la dose est intégrée dans l'alimentation, le composé à tester doit être distribué uniformément dans la ration.
vii) Si, par suite du métabolisme ou d'autres processus se produisant dans ou sur les végétaux traités, ou, par suite de la transformation de produits traités, le résidu final (auquel les consommateurs ou les travailleurs visées à l'annexe II, point 7.2.3 sont exposés) contient une substance qui n'est pas la substance active proprement dite et qui n'est pas identifiée comme un métabolite chez les mammifères, il est nécessaire d'effectuer des études de toxicité sur ces composants du résidu final sauf s'il peut être démontré que l'exposition du consommateur ou du travailleur à ces substances ne présente pas un risque important pour la santé. Des études de toxicocinétique et de métabolisme se rapportant aux métabolites et aux produits cataboliques ne doivent être effectuées que si la toxicité du métabolite ne peut pas être déduite des résultats disponibles se rapportant à la substance active.
viii) Le mode d'administration de la substance d'essai dépend des principaux types d'exposition. Si l'exposition est esentiellement une exposition à la phase gazeuse, il peut être préférable de réaliser des études par voie inhalatoire au lieu d'études par voie orale.
5.1. Etudes de l'absorption, de la distribution, de l'excrétion et du métabolisme chez les mammifères.
Il est possible que les seules données requises à cet effet soient des données très limitées, décrites ci-dessous, et portant sur une seule espèce d'essai (normalement le rat). Ces données peuvent fournir des informations utiles pour la conception et l'interprétation des essais de toxicité ultérieurs.
Cependant, il convient de se rappeler que les informations relatives aux différences entre les espèces peuvent être déterminantes pour l'extrapolation à l'homme des données relatives à l'animal, et les informations sur la pénétration cutanée, l'absorption, la distribution, l'excrétion et le métabolisme devraient être utiles pour l'évaluation du risque pour l'opérateur. Il est impossible de préciser les exigences détaillées concernant les informations dans tous les domaines étant donné que les exigences précises dépendent des résultats obtenus pour chaque substance d'essai particulière.
But des essais :
Les essais doivent fournir des données suffisantes pour permettre :
- une évaluation du taux et de l'importance de l'absorption ;
- une évaluation de la distribution dans les tissus et du taux ainsi que de l'importance de l'excrétion de la substance à tester et des métabolites importants ;
- l'identification des métabolites et du schéma métabolique.
Il convient aussi de rechercher l'effet de la dose sur ces paramètres et de déterminer si les résultats sont différents après l'administration d'une dose unique ou de doses répétées.
Situations dans lesquelles les essais sont requis :
Une étude toxicocinétique à dose unique sur des rats (voie d'administration orale) pour au moins deux concentrations ainsi qu'une étude toxicocinétique à doses répétées, à une seule concentration, sur des rats (voie d'administration orale) doivent être réalisées et relatées. Il peut être nécessaire, dans certains cas, de procéder à des études complémentaires sur une autre espèce (par exemple la chèvre ou le poulet).
Ligne directrice pour l'essai :
Directive 88/302/CEE de la Commission du 18 novembre 1987 portant neuvième adaptation au progrès technique de la directive 67/548/CEE du Conseil concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à la classification, l'emballage et l'étiquetage des substances dangereuses, partie B, Toxicocinétique.
5.2. Toxicité aiguë.
Les études, données et informations à fournir et à évaluer doivent être suffisantes pour permettre de déceler les effets d'une exposition unique à la substance active et en particulier d'établir ou d'indiquer :
- la toxicité de la substance active :
- l'évolution au cours du temps et les caractéristiques des effets avec des détails exhaustifs sur les modifications comportementales et les éventuelles constatations macro-pathologiques à l'inspection post mortem :
- si possible, le mode d'action toxique ;
- le danger relatif lié à diverses voies d'exposition.
Si l'accent doit être mis sur l'estimation des degrés de toxicité en cause, les informations obtenues doivent aussi permettre de classifier la substance active conformément à la directive 67/548/CEE du Conseil. Les informations obtenues grâce à un essai de toxicité aiguë revêtent une importance particulière pour l'évaluation des dangers potentiels en cas d'accident.
5.2.1. Toxicité orale.
Situations dans lesquelles l'essai est requis.
La toxicité orale aiguë de la substance active doit toujours être relatée.
Ligne directrice pour l'essai :
L'essai doit être effectué conformément à l'annexe de la directive 92/69/CEE de la Commission du 31 juillet 1992 portant dix-septième adaptation au progrès technique de la directive 67/548/CEE du Conseil concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à la classification, l'emballage et l'étiquetage des substances dangereuses, méthode B 1 ou B 1 bis.
5.2.2. Toxicité dermale.
Situations dans lesquelles l'essai est requis :
La toxicité dermale aiguë de la substance active doit toujours être relatée.
Ligne directrice pour l'essai :
Les effets locaux et systémiques doivent être analysés. L'essai doit être effectué conformément à la directive 92/69/CEE, méthode B 3.
5.2.3. Toxicité inhalatoire.
Situations dans lesquelles l'essai est requis.
La toxicité par inhalation de la substance active doit être relatée si cette dernière :
- est un gaz, notamment liquéfié ;
- doit être utilisée comme fumigant ;
- doit être incorporée dans une préparation fumigène, un aérosol ou produisant de la vapeur ;
- doit être utilisée à l'aide d'un équipement de nébulisation ;
- a une pression de vapeur 1 x 10-2 Pa et doit être incorporée dans des préparations à utiliser dans des espaces clos tels que des magasins ou des serres ;
- doit être incorporée dans des préparations poudreuses contenant une proportion significative de particules d'un diamètre 50 micro m (1 p. 100 sur la base du poids) ;
- doit être incorporée dans des préparations à appliquer selon un procédé produisant une proportion significative de particules ou de gouttelettes d'un diamètre 50 micro m (1 p. 100 sur la base du poids).
Ligne directrice pour l'essai :
L'essai doit être effectué conformément à la directive 92/69/CEE, méthode B 2.
5.2.4. Irritation de la peau.
But de l'essai :
L'essai doit permettre de mettre en évidence le pouvoir irritant pour la peau de la substance active, y compris la réversibilité éventuelle des effets observés.
Situations dans lesquelles l'essai est requis :
Le pouvoir irritant pour la peau de la substance active doit être déterminé sauf si, comme il est indiqué dans la ligne directrice pour l'essai, il est probable que des effets graves sur la peau peuvent se produire ou que ces effets peuvent être exclus.
Ligne directrice pour l'essai :
L'essai relatif à l'irritation aiguë de la peau doit être effectué conformément à la directive 92/69/CEE, méthode B 4.
5.2.5. Irritation des yeux.
But de l'essai :
L'essai doit permettre de mettre en évidence le pouvoir irritant pour les yeux de la substance active, y compris la réversibilité potentielle des effets observés.
Situations dans lesquelles l'essai est requis :
L'essai relatif à l'irritation pour les yeux doit être effectué sauf si, comme il est indiqué dans la ligne directrice pour l'essai, il est probable que des effets graves sur les yeux peuvent se produire.
Ligne directrice pour l'essai :
L'irritation aiguë des yeux doit être déterminée conformément à la directive 92/69/CEE, méthode B 5.
5.2.6. Sensibilisation de la peau.
But de l'essai :
L'essai doit fournir des données suffisantes pour permettre une évaluation de la capacité de la substance active de provoquer des réactions de sensibilisation de la peau.
Situations dans lesquelles l'essai est requis :
L'essai doit être réalisé en toute circonstance sauf si la substance est un sensibilisant connu.
Ligne directrice pour l'essai :
L'essai doit être réalisé conformément à la directive 92/69/CEE, méthode B 6.
5.3. Toxicité à court terme.
Les études de toxicité à court terme doivent être conçues pour fournir des informations sur la quantité de substance active pouvant être tolérée sans provoquer d'effets toxiques dans les conditions de l'étude. De telles études fournissent des données utiles sur les risques encourus par des personnes manipulant et utilisant des préparations contenant la substance active. En particulier, les études à court terme donnent un aperçu déterminant des effets cumulés possibles de la substance active et des risques encourus par les travailleurs exposés de façon intensive. En outre, les études à court terme donnent des informations utiles pour la conception des études de toxicité chronique.
Les études, données et informations à fournir et à évaluer doivent être suffisantes pour permettre de déceler les effets découlant d'une exposition répétée à la substance active, et, en outre, d'établir ou d'indiquer notamment :
- la relation entre la dose et les effets néfastes ;
- la toxicité de la substance active, y compris, si possible, le NOAEL ;
- les organes cibles, si pertinent ;
- l'évolution au cours du temps et les caractéristiques de l'empoisonnement avec des détails exhaustifs sur les modifications comportementales et les éventuelles constatations macropathologiques à l'inspection post mortem ;
- les effets toxiques particuliers et les changements pathologiques produits ;
- le cas échéant, la persistance et la réversibilité de certains effets toxiques observés, à la suite d'une interruption d'administration ;
- si possible, le mode d'action toxique ;
- le danger relatif lié à diverses voies d'exposition.
5.3.1. Etude de 28 jours par voie orale.
Situations dans lesquelles l'essai est requis :
Bien qu'il ne soit pas obligatoire d'effectuer des études à court terme de 28 jours, celles-ci peuvent être utiles à titre d'essais d'orientation. S'ils sont effectués, il convient de les relater, étant donné que leurs résultats pourraient avoir une valeur particulière pour l'identification des réactions d'adaptation qui peuvent être masquées dans des études de toxicité chronique.
Ligne directrice pour l'essai :
L'essai doit être réalisé conformément à la directive 92/69/CEE, méthode B 7.
5.3.2. Etude de 90 jours par voie orale.
Situations dans lesquelles l'essai est requis :
La toxicité orale à court terme (90 jours) de la substance active pour le rat et le chien doit être relatée en toute circonstance. S'il s'avère que le chien est beaucoup plus sensible et si les données obtenues peuvent présenter un intérêt en vue de l'extrapolation des résultats obtenus à l'homme, une étude de toxicité de 12 mois sur les chiens doit être effectuée et relatée.
Lignes directrices pour l'essai :
Directive 88/302/CEE, partie B, essai de toxicité orale subchronique.
5.3.3. Autres voies.
Situations dans lesquelles l'essai est requis :
Des études supplémentaires sur la toxicité par voie cutanée peuvent être utiles pour l'évaluation de l'exposition de l'opérateur.
Pour les substances volatiles (pression de vapeur 10-2 pascal), un jugement d'expert sera requis afin de décider si les études à court terme doivent être réalisées par voie orale ou inhalatoire.
Lignes directrices pour l'essai :
- étude de toxicité dermique de 28 jours : directive 92/69/CEE, méthode B 9 ;
- étude de toxicité dermique de 90 jours : directive 88/302/CEE, partie B, étude de toxicité dermique subchronique.
- étude de toxicité de 28 jours par inhalation : directive 92/69/CEE, méthode B 8 ;
- étude de toxicité de 90 jours par inhalation : directive 88/302/CEE, partie B, étude de toxicité subchronique par inhalation.
5.4. Essais de génotoxicité.
But de l'essai.
Ces études présentent un intérêt pour :
- la prédiction du pouvoir génotoxique ;
- l'identification précoce des cancérogènes génotoxiques ;
- l'explication du mécanisme d'action de certains cancérogènes.
Pour éviter toute réponse qui serait le résultat d'artefacts du système d'essai, il faut éviter d'utiliser des doses excessivement toxiques dans les essais de mutagenèse in vitro ou in vivo. Cette procédure doit être considérée comme une orientation générale. Il importe d'adopter une attitude souple, les autres tests à réaliser devant être fonction de l'interprétation des résultats à chaque étape.
5.4.1. Etudes in vitro.
Situations dans lesquelles les essais sont requis :
Des essais de mutagenèse in vitro (essai bactérien relatif à la mutation génique, essai de clastogénicité dans des cellules de mammifères et essai de mutation génique dans des cellules de mammifères) doivent toujours être réalisés.
Lignes directrices pour les essais.
Des exemples d'essais acceptables sont notamment les suivants :
- directive 92/69/CEE, méthode B 14, essai de mutation reverse sur Salmonella typhimurium ;
- directive 92/69/CEE, méthode B 10, essai de cytogénétique in vitro sur mammifère ;
- directive 88/302/CEE, partie B, cellules de mammifère in vitro, essai de mutation génique.
5.4.2. Etudes in vitro sur cellules somatiques.
Situations dans lesquelles les essais sont requis :
Si tous les résultats des études in vitro sont négatifs, d'autres tests devront être réalisés en tenant compte d'autres informations pertinentes disponibles (y compris des données toxicocinétiques, toxicodynamiques, physicochimiques et données sur des substances analogues). Ces études pourraient être une étude in vivo ou une étude in vitro avec un système métabolique différent de celui ou ceux utilisés auparavant.
Si l'essai cytogénétique in vitro est positif, il faut effectuer un essai in vivo sur des cellules somatiques (analyse de métaphases des cellules de la moelle osseuse de rongeur ou essai du micronoyau chez les rongeurs).
Si l'un ou l'autre des essais de mutation génique in vitro est positif, il faut effectuer un essai in vivo afin d'analyser la synthèse non programmée d'ADN ou un "spot test" chez la souris.
Lignes directrices pour les essais :
Des exemples de lignes directrices acceptables pour les essais sont les suivants :
- directive 92/69/CEE, méthode B 12, test du micronoyau ;
- directive 88/302/CEE, partie B, "spot test" chez la souris ;
- directive 92/69/CEE, méthode B 11, essai de cytogénétique in vivo sur la moelle osseuse de mammifère, analyse chromosomique.
5.4.3. Etudes in vivo sur cellules germinales.
Situations dans lesquelles les essais sont requis :
Si l'un quelconque des résultats des essais effectués in vitro sur cellules somatiques est positif, la réalisation d'un essai in vivo permettant de déterminer les effets sur les cellules germinales peut être justifiée. La nécessité d'effectuer ces essais doit être examinée au cas par cas compte tenu des informations concernant la toxicocinétique, l'utilisation et l'exposition probable. Des essais appropriés devront permettre d'examiner l'interaction avec l'ADN (tels que l'essai de létalité dominante), de déterminer la possibilité de développer d'effets héréditaires et si possible de les estimer quantitativement. Il est reconnu que, étant donné leur complexité, l'utilisation d'études quantitatives supposerait une justification solidement fondée.
5.5. Toxicité à long terme et cancérogenèse.
Buts de l'essai.
Les études à long terme effectuées et relatées, prises en compte avec d'autres données et informations importantes concernant la substance active, doivent être suffisantes pour permettre de déceler les effets résultant d'expositions répétées à la substance active et être suffisantes notamment pour :
- identifier les effets néfastes résultant de l'exposition à la substance active ;
- identifier les organes cibles, si pertinents ;
- établir la relation dose-réponse ;
- identifier les changements dans les signes et les manifestations de toxicité observés ;
- fixer le NOAEL.
De même, les études de cancérogenèse considérées avec d'autres données et informations pertinentes sur la substance active doivent être suffisantes pour permettre d'évaluer les dangers pour l'homme ayant subi des expositions répétées à la substance active, et en particulier doivent être suffisantes :
- pour identifier les effets cancérogènes résultant de l'exposition à la substance active ;
- déterminer les espèces et la spécificité organique des tumeurs induites ;
- établir la relation dose-réponse ;
- pour les cancérogènes non génotoxiques, identifier la dose maximale sans effet néfaste (dose seuil).
Situations dans lesquelles l'essai est requis :
La toxicité à long terme et la cancérogenèse de toute substance active doivent être déterminées. Si, dans des situations exceptionnelles, il est déclaré que de tels essais ne sont pas nécessaires, ces déclarations doivent être pleinement justifiées, par exemple lorsque des données toxicocinétiques prouvent que l'absorption de la substance active ne se fait pas dans le tractus digestif, par la peau ou le système respiratoire.
Conditions d'essai :
Une étude de toxicité à long terme et une étude de cancérogenèse par voie orale (deux ans) relative à la substance active doivent être effectuées sur le rat ; ces études peuvent être combinées.
Une étude de cancérogenèse de la substance active doit aussi être effectuée sur la souris.
Si un mécanisme de cancérogenèse non génotoxique est supposé, un dossier bien argumenté, étayé de données expérimentales pertinentes, y compris celles nécessaires pour expliquer les mécanismes éventuellement en cause, doit être fourni.
Si les points de référence types pour les réactions au traitement sont des données provenant de contrôles simultanés, des données de contrôles historiques peuvent être utiles dans l'interprétation de certaines études de cancérogenèse. Si elles sont présentées, les données de contrôle historiques devraient concerner la même espèce et la même souche d'animaux maintenus dans des conditions similaires, et provenir d'études faites à la même époque. Les informations sur les données de contrôle historiques fournies doivent comprendre :
- l'identification de l'espèce et de la souche, le nom du fournisseur et l'identification de la colonie spécifique si le fournisseur est implanté dans plusieurs sites géographiques ;
- le nom du laboratoire et les dates auxquelles l'étude a été réalisée ;
- la description des conditions générales dans lesquelles les animaux ont été maintenus, y compris le type ou la marque de la ration alimentaire et, si possible, les quantités consommées ;
- l'âge approximatif, exprimé en jours, des animaux témoins au début de l'étude et à la date de sacrifice des animaux ou de leur mort ;
- la description du schéma de mortalité du groupe témoin constaté pendant ou à la fin de l'étude et d'autres observations pertinentes (par exemple, maladies, infections) ;
- le nom du laboratoire et des experts scientifiques chargés de la réalisation de l'étude et de la collecte et de l'interprétation des données pathologiques relatives à l'étude ;
- une déclaration relative à la nature des tumeurs qui peuvent avoir été combinées pour produire une quelconque des données d'incidence.
Les doses expérimentées, y compris la dose la plus élevée, doivent être sélectionnées sur la base des résultats d'essais à court terme et, si elles sont disponibles, à la date de programmation des études considérées sur la base des données de métabolisme et de la toxicocinétique. La dose la plus élevée appliquée dans l'étude de la cancérogénicité devrait produire des signes de toxicité minimale, telle qu'une légère atténuation du gain de poids corporel (moins de 10 p. 100), sans provoquer de nécrose tissulaire ou de saturation métabolique ni d'altération substantielle de la durée de vie normale due à des effets autres que des tumeurs. Si l'étude de toxicité à long terme est effectuée séparément, la dose la plus élevée devrait produire des signes évidents de toxicité sans provoquer une létalité excessive. Des doses plus élevées, produisant une toxicité excessive, ne sont pas considérées comme pertinentes pour les évaluations à effectuer.
Dans la collecte des données et la compilation des rapports, l'incidence des tumeurs bénignes et celle des tumeurs malignes ne doivent pas être combinées, sauf s'il existe une preuve évidente que les tumeurs bénignes se transforment en tumeurs malignes avec le temps. De même, des tumeurs dissemblables, non associées, bénignes ou malignes, apparaissant dans le même organe, ne doivent pas être combinées pour l'établissement des rapports. Pour prévenir toute confusion, une terminologie telle que celle mise au point par l'American Society of Toxicologic Pathologists (1) ou le Hannover Tumour Registry (RENI) devrait être utilisée dans la nomenclature et l'établissement des rapports concernant les tumeurs. Le système utilisé doit être identifié.
(1) Standardized System of Nomenclature and Diagnostic Criteria - Guides for Toxicologic Pathology.
Il est d'une importance capitale que le matériel biologique retenu pour l'examen histopathologique comprenne du matériel sélectionné pour donner d'autres informations sur les lésions constatées au cours de l'examen macropathologique. Si elles conviennent pour élucider le mécanisme d'action et si elles sont disponibles, des techniques histologiques spécifiques (coloration), des techniques histochimiques et des examens au microscope électronique doivent être effectués et relatés.
Ligne directrice pour le test :
Les études doivent être effectuées conformément à la directive 88/302/CEE, partie B, étude de la toxicité chronique, étude de la cancérogénèse ou étude combinée de toxicité chronique et de cancérogénèse.
5.6. Test de reproduction.
Les effets néfastes pour la reproduction sont de deux types :
- troubles de la fertilité mâle ou femelle ;
- effets sur le développement normal de la descendance (toxicité du développement).
Les effets possibles sur les aspects physiologiques de la reproduction tant chez les mâles que chez les femelles ainsi que les effets éventuels sur le développement prénatal et postnatal doivent être recherchés et relatés. Si, dans des situations exceptionnelles, ces essais sont déclarés superflus, cette affirmation doit être entièrement justifiée.
Si les points de référence types pour les réponses au traitement sont des données provenant de contrôles simultanés, des données de contrôles historiques peuvent être utiles dans l'interprétation de certaines études de reproduction particulières. Si elles sont présentées, les données de contrôle historiques devraient concerner la même espèce et la même souche d'animaux, maintenus dans des conditions similaires, et devraient provenir d'études faites à la même époque. Les informations concernant les données de contrôle historiques doivent comporter :
- l'identification de l'espèce et de la souche, le nom du fournisseur et l'identification de la colonie spécifique si le fournisseur est implanté dans plusieurs sites géographiques ;
- le nom du laboratoire et les dates auxquelles l'étude a été réalisée ;
- la description des conditions générales dans lesquelles les animaux ont été maintenus, y compris le type ou la marque de la ration alimentaire et, si possible, les quantités consommées ;
- l'âge approximatif, exprimé en jours, des animaux témoins au début de l'étude et à la date de sacrifice des animaux ou de leur mort ;
- la description du schéma de mortalité du groupe témoin constaté pendant ou à la fin de l'étude et d'autres observations pertinentes (par exemple, maladies, infections) ;
- le nom du laboratoire et des experts scientifiques chargés de la réalisation de l'étude et de la collecte et de l'interprétation de données toxicologiques relatives à l'étude.
5.6.1. Etudes sur plusieurs générations.
But de l'essai :
Les études relatées, considérées avec d'autres données et informations importantes sur la substance active, doivent être suffisantes pour permettre de déceler les effets pour la reproduction découlant d'une exposition répétée à la substance active et doivent notamment être suffisantes pour :
- identifier les effets directs et indirects sur la reproduction d'une exposition à la substance active ;
- identifier le taux d'accroissement des effets toxiques globaux (constatés lors des essais de toxicité chronique et à court terme) ;
- fixer la relation dose-réponse ;
- identifier les changements dans les signes et manifestations de toxicité observés ;
- fixer le NOAEL.
Situations dans lesquelles l'essai est requis :
Une étude de reproduction sur deux générations chez les rats doit toujours être relatée.
Ligne directrice pour l'essai :
Les essais doivent être effectués conformément à la directive 88/302/CEE, partie B, test de reproduction sur deux générations. En outre les poids des organes de reproduction doit être relaté.
Etudes supplémentaires :
Pour obtenir une meilleure interprétation des effets sur la reproduction et dans la mesure où cette information n'est pas encore disponible, il pourrait être utile de procéder à des études supplémentaires afin de relater les informations sur les points suivants :
- études séparées pour les mâles et les femelles ;
- études en trois étapes ("segment") ;
- test de létalité dominante pour la fertilité mâle ;
- accouplements croisés de mâles traités avec des femelles non traitées et vice-versa ;
- effet sur la spermatogénèse ;
- effets sur l'ovogenèse ;
- motilité, mobilité et morphologie des spermatozoïdes ;
- étude de l'activité hormonale.
5.6.2. Etudes de développement.
But des essais :
Les études relatées considérées en même temps que d'autres données et informations pertinentes sur la substance active doivent être suffisantes pour permettre d'évaluer les effets sur le développement de l'embryon et du foetus, à la suite d'expositions répétées à la substance active et doivent notamment être suffisantes pour :
- identifier les effets directs et indirects sur le développement de l'embryon et du foetus à la suite d'une exposition à la substance active ;
- identifier toute toxicité chez la mère ;
- établir la relation entre les réponses observées et la dose tant chez la femelle que dans sa descendance ;
- identifier les changements de signes et de manifestations de toxicité observés ;
- fixer le NOAEL.
Par ailleurs, les essais donneront des informations supplémentaires sur toute aggravation des effets toxiques globaux chez les animaux gravides.
Situations dans lesquelles l'essai est requis.
Les essais doivent toujours être effectués.
Conditions d'essai :
La toxicité pour le développement doit être déterminée chez le rat et chez le lapin après une exposition par voie orale. Noter séparément les malformations et les altérations. Un glossaire terminologique et les principes de diagnostic doivent être donnés dans le rapport pour toutes les malformations et altérations.
Ligne directrice pour les essais :
Les essais doivent être effectués conformément à la directive 88/302/CEE, partie B, étude de tératogénicité.
5.7. Etudes de neurotoxicité retardée.
But de l'essai :
L'essai doit fournir des données suffisantes pour examiner si la substance active pourrait provoquer une neurotoxicité différée après exposition aiguë.
Situations dans lesquelles les essais sont requis :
Ces études doivent être effectuées pour les substances de structure analogue ou apparentée à la structure de celles susceptibles d'induire une neurotoxicité retardée comme les organophosphates.
Ligne directrice pour les essais :
Les essais doivent être effectués conformément à la ligne directrice 418 de l'O.C.D.E.
5.8. Autres études toxicologiques.
5.8.1. Etudes de toxicité des métabolites visés au point vii de l'introduction.
Les études complémentaires concernant des substances autres que la substance active ne sont pas requises de façon routinière.
Les décisions relatives à la nécessité d'effectuer des études complémentaires doivent être prises cas par cas.
5.8.2. Etudes complémentaires sur la substance active.
Dans certains cas, il peut être nécessaire d'effectuer des études complémentaires pour clarifier certains effets observés. Ces études pourraient comprendre :
- des études sur l'absorption, la distribution, l'excrétion et le métabolisme ;
- des études sur le potentiel neurotoxique ;
- des études sur le potentiel immunotoxicologique ;
- des études par d'autres voies d'administration.
Les décisions relatives à la nécessité d'effectuer des études complémentaires doivent être prises cas par cas, compte tenu des résultats des études toxicologiques et de métabolisme existants et des types d'exposition les plus importants.
Les études requises peuvent être conçues sur une base individuelle, compte tenu des paramètres spécifiques à examiner et des objectifs à atteindre.
5.9. Données médicales.
Si elles sont disponibles, et sans préjudice des dispositions de l'article 5 de la directive 80/1107/CEE du Conseil du 27 novembre 1980 concernant la protection des travailleurs contre les risques liés à une exposition à des agents chimiques, physiques et biologiques pendant le travail, les données et informations pratiques importantes pour la reconnaissance des symptômes d'empoisonnement et sur l'efficacité des premiers soins et mesures thérapeutiques doivent être présentées. Des références plus spécifiques concernant l'étude sur animaux de la pharmacologie relative aux antidotes ou à la sécurité doivent être fournies. S'il y a lieu, l'efficacité d'antidotes potentiels à l'empoisonnement doit être étudiée et relatée.
Les données et informations importantes pour les effets de l'exposition de l'homme, si elles sont disponibles et ont la qualité nécessaire, ont une valeur particulière parce que confirmant le bien-fondé des extrapolations faites et des conclusions relatives aux organes cibles, aux relations doses-réponses et à la réversibilité des effets toxiques. De telles données peuvent être obtenues après une exposition accidentelle ou professionnelle.
5.9.1. Surveillance médicale du personnel de l'usine de production.
Des rapports relatifs aux programmes de surveillance de la santé du personnel, étayés d'informations détaillées sur la conception du programme, l'exposition à la substance active et l'exposition à d'autres produits chimiques, doivent être présentés. De tels rapports doivent, si possible, comprendre des données pertinentes du point de vue mécanisme d'action de la substance active. Ces rapports doivent éventuellement comporter si elles sont disponibles des données relatives aux personnes exposées dans les usines de production ou après application de la substance active (par exemple dans des essais d'efficacité).
Les informations disponibles sur la sensibilisation, y compris la réponse allergique des travailleurs et autres personnes exposées à la substance active doivent être fournies et comporter, le cas échéant, des informations relatives à toute incidence d'hypersensibilité. Les informations fournies doivent comporter des détails sur la fréquence, le niveau et la durée de l'exposition, les symptômes observés et autres informations cliniques pertinentes.
5.9.2. Observation directe, par exemple cas clinique et cas d'empoisonnement.
Les rapports disponibles provenant de sources bibliographiques publiques, concernant des cas cliniques et des cas d'empoisonnement doivent, s'ils sont empruntés à des revues autorisées ou à des rapports officiels, être présentés avec les rapports de toutes les études de suivi entreprises. Ces rapports doivent comporter des descriptions exhaustives de la nature, du degré et de la durée de l'exposition ainsi que les symptômes cliniques observés, les dispositions relatives aux premiers soins et mesures thérapeutiques appliqués ainsi que les données mesurées et les observations faites. Un résumé ou des informations succinctes sont sans intérêt.
Si elle est étayée de précisions suffisantes, une telle documentation peut présenter une valeur particulière pour confirmer la validité des extrapolations à l'homme à partir de données relatives à l'animal et pour identifier des effets néfastes imprévus particuliers à l'homme.
5.9.3. Observations sur l'exposition de la population en général et, le cas échéant, études épidémiologiques.
Lorsqu'elles existent et qu'elles sont étayées par des données sur les degrés et la durée d'exposition, et réalisées conformément à des normes (2) reconnues, des études épidémiologiques présentent un intérêt particulier et doivent être soumises.
(2) Guidelines for Good Epidemiology Practices for Occupational and Environmental Research, developed by the Chemical Manufactures Association's Epidemiology Task Group, as part of the Epidemiology Resource and Information Centre (ERIC), Pilot Project, 1991.
5.9.4. Diagnostic de l'empoisonnement (détermination de la substance active, de métabolites), signes spécifiques d'empoisonnement, tests cliniques.
Une description détaillée des signes et symptômes cliniques d'empoisonnement, y compris les signes et symptômes précoces ainsi que les détails complets des tests cliniques utiles pour des fins diagnostiques, doit, le cas échéant, être fournie et comporter des informations détaillées sur l'évolution au cours du temps concernant l'ingestion, l'exposition cutanée ou l'inhalation de diverses quantités de la substance active.
5.9.5. Traitement proposé : premiers soins, antidotes, traitement médical.
Les premiers soins à appliquer en cas d'empoisonnement (réel ou supposé) et en cas de contamination des yeux doivent être prévus.
Les traitements thérapeutiques à appliquer en cas d'empoisonnement ou de contamination des yeux, y compris, éventuellement, l'utilisation d'antidote, doivent faire l'objet d'une description détaillée. Les informations fondées sur l'expérience pratique, éventuellement disponible, et, dans d'autres cas, sur des considérations théoriques, telles que l'efficacité de traitements thérapeutiques de remplacement, doivent être fournies le cas échéant. Les contre-indications liées à certains traitements, particulièrement ceux touchant les "problèmes médicaux généraux" et les conditions doivent être décrites.
5.9.6. Effets prévisibles d'un empoisonnement.
S'ils sont connus, les effets prévisibles d'un empoisonnement et la durée de ceux-ci doivent être décrits et comprendre :
- l'impact du type, du niveau et de la durée de l'exposition ou de l'ingestion ;
- les laps de temps variables entre l'exposition ou l'ingestion et le commencement du traitement.
5.10. Résumé de la toxicologie chez les mammifères et évaluation globale.
Un résumé de toutes les données et informations fournies en application des points 5.1 à 5.10 doit être présenté et doit comporter une évaluation détaillée et critique desdites données sur la base de critères et de lignes directrices pertinentes concernant l'évaluation et la prise de décision, compte tenu particulièrement des risques potentiels ou effectifs pour l'homme et les animaux ainsi que de l'ampleur, de la qualité et de la fiabilité de la base de données.
Le cas échéant, compte tenu du profil analytique des lots de la substance active (point 1.11) et de toutes études complémentaires effectuées (point 5 iv), la pertinence des données proposées pour l'évaluation du profil toxicologique de la substance fabriquée doit être étayée.
A partir d'une évaluation de la base de données ainsi que des critères et lignes directrices pertinents pour la décision, des justifications doivent être données pour les NOAEL proposés pour chaque étude pertinente.
Sur la base de ces données, des propositions fondées scientifiquement, relatives à la fixation d'une DJA, d'un NAEO (de NAEO) concernant la substance active, doivent être présentées.
6. Résidus dans ou sur les produits traités, les denrées alimentaires et les aliments pour animaux.
(Arrêté du 1er février 1996, article 1er et Arrêté du 1er décembre 1998, article 1er II)
Introduction.
i) Les informations fournies, considérées avec celles données pour une ou plusieurs préparations contenant la substance active, doivent être suffisantes pour permettre une évaluation des risques pour l'homme provenant des résidus de la substance active et des métabolites pertinents, produits de dégradation et de réaction restant dans l'aliment. En outre, les informations fournies doivent être suffisantes pour :
- permettre d'arrêter une décision relative à la possibilité d'inclusion de la substance active dans la liste communautaire des substances actives ;
- préciser les conditions appropriées ou restrictions à prévoir pour toute inclusion dans la liste communautaire des substances actives.
ii) Une description détaillée (spécification) de la substance utilisée, visée au point 1.11 doit être fournie.
iii) Les études devraient être effectuées conformément aux instructions disponibles pour les procédures d'analyse réglementaires des résidus phytopharmaceutiques contenus dans les denrées alimentaires (1).
(1) Instructions en cours d'élaboration.
iv) Le cas échéant, les données sont analysées à l'aide de méthodes statistiques appropriées. Donner des informations complètes sur l'analyse statistique.
v) Stabilité des résidus pendant le stockage :
Il peut être nécessaire d'effectuer des études sur la stabilité des résidus pendant le stockage. Sauf si un composé est réputé par ailleurs volatil ou instable, les données ne sont pas requises pour les échantillons extraits et analysés dans les trente jours à compter du prélèvement des échantillons (six mois s'il s'agit d'une substance radiomarquée), pourvu que ceux-ci soient généralement congelés dans les vingt-quatre heures suivant le prélèvement des échantillons ;
Des études au moyen de substances on radiomarquées doivent être effectuées avec des substrats représentatifs et, de préférence, sur des échantillons provenant de cultures ou d'animaux traités contenant des résidus. En cas d'impossibilité, en revanche, des fractions aliquotes d'échantillons de contrôle préparés devraient être additionnées d'une quantité connue de produit chimique avant d'être entreposées dans des conditions de stockage normales ;
Si la dégradation au cours du stockage est significative (plus de 30 %), il peut être nécessaire de modifier les conditions de stockage ou de ne pas stocker les échantillons avant analyse et de répéter les études dans lesquelles les conditions de stockage n'étaient pas satisfaisantes ;
Il convient de présenter des informations détaillées sur la préparation de l'échantillon et les conditions de stockage (température et durée) des échantillons et extraits. Les données concernant la stabilité au stockage sur la base d'extraits d'échantillons devront aussi être exigées sauf si les échantillons sont analysés dans un délai de vingt-quatre heures après leur extraction.
6.1. Métabolisme, distribution et expression du résidu dans les végétaux.
But des essais.
Les objectifs de ces études sont les suivants :
- permettre une estimation des résidus finaux totaux dans la fraction pertinente des produits de la récolte qui ont été traités selon le programme prévu ;
- identifier les composants principaux du résidu final total ;
- indiquer la distribution des résidus entre les fractions pertinentes du produit de la récolte ;
- quantifier les composants principaux du résidu et établir l'efficacité des méthodes d'extraction de ces composants ;
- fixer la définition et l'expression d'un résidu.
Situations dans lesquelles les essais sont requis.
Ces études doivent toujours être effectuées sauf s'il est possible de prouver qu'aucun résidu ne reste sur les végétaux/produits végétaux utilisés comme denrées alimentaires ou aliments pour animaux.
Conditions des essais.
Les études de métabolisme doivent porter sur des cultures ou catégories de culture dans lesquelles seraient utilisés des produits phytopharmaceutiques contenant la substance active.
Si l'on considère une vaste gamme d'utilisation sur diverses catégories de cultures ou dans la catégorie des fruits, il convient d'effectuer des études sur au moins trois cultures sauf s'il peut être justifié qu'un métabolisme différent est peu probable. Dans les cas où l'utilisation est prévue sur diverses catégories de cultures, les études doivent être représentatives des catégories visées. A cet effet, les cultures peuvent être considérées comme appartenant à une des cinq catégories suivantes : légumes racines, cultures à feuilles, fruits, légumineuses et oléagineux, céréales. Si des études existent pour les cultures appartenant à trois de ces catégories et que les résultats démontrent que le mode de dégradation est similaire pour l'ensemble de ces trois catégories, il est peu probable que des études complémentaires soient nécessaires, sauf si l'on peut s'attendre à un métabolisme différent. Les études du métabolisme doivent aussi prendre en compte les diverses propriétés de la substance active et la méthode d'application prévue.
Evaluation des résultats des diverses études à donner au sujet du point et de la voie d'absorption (par exemple par les feuilles ou les racines) et de la distribution des résidus entre les parties caractéristiques de la culture au moment de la récolte (l'accent étant placé en particulier sur les parties pouvant être utilisées pour l'alimentation humaine ou animale). Si la substance active ou les métabolites pertinents ne sont pas absorbés par la culture, en donner l'explication. Les informations sur le mode d'action et les propriétés physico-chimiques de la substance active peuvent être utilisées pour interpréter les données des essais.
6.2. Métabolisme, distribution et expression des résidus dans les animaux d'élevage.
But des essais.
Les objectifs de ces études sont les suivants :
- identifier les principaux composants du résidu final total dans les produits animaux comestibles :
- chiffrer le taux de dégradation et d'excrétion du résidu total dans certains produits animaux (lait ou œufs) et excrétions animales ;
- indiquer la distribution des résidus entre les produits animaux comestibles pertinents ;
- quantifier les principaux composants du résidu et démontrer l'efficacité des méthodes d'extraction de ces composants ;
- établir des données pouvant servir à la prise d'une décision sur la nécessité d'effectuer des études sur l'alimentation animale conformément au point 6.4 ;
- prendre une décision quant à la définition et l'expression d'un résidu.
Situations dans lesquelles les essais sont requis.
Les études du métabolisme sur des animaux, tels que les ruminants en lactation (par exemple la chèvre ou la vache) ou la volaille en période de ponte, ne sont requises que si l'utilisation du pesticide peut aboutir à la constitution de taux de résidus significatifs dans les aliments pour animaux (0,1 mg/kg de la ration fourragère totale ingérée, sauf cas spéciaux concernant par exemple des substances actives accumulables). S'il apparaît que les voies du métabolisme diffèrent dans des proportions significatives chez le rat par rapport aux ruminants, une étude sur le porc doit être effectuée sauf si l'on prévoit que l'absorption par les porcs ne sera pas significative.
6.3. Essais relatifs aux résidus.
But des essais.
Les objectifs de ces études sont les suivants :
- quantifier les concentrations de résidus maximales probables contenues dans les cultures traitées, au moment de la récolte ou de la sortie du stock conformément aux bonnes pratiques agricoles proposées, et
- déterminer, le cas échéant, le rythme de diminution des dépôts du produit phytopharmaceutique.
Situations dans lesquelles les essais sont requis.
Ces études doivent toujours être effectuées lorsque le produit phytopharmaceutique doit être appliqué sur des végétaux/produits végétaux utilisés comme denrées alimentaires ou aliments pour les animaux ou lorsque les résidus contenus dans le sols ou dans d'autres substrats peuvent être absorbés par ces végétaux, sauf s'il est possible de pratiquer une extrapolation à partir de données adéquates sur une autre culture.
Les données d'essais relatifs aux résidus seront proposées dans le dossier de l'annexe I pour les utilisations de produits phytopharmaceutiques pour lesquels l'autorisation est demandée à la date d'introduction d'un dossier d'insertion de la substance active dans la liste communautaire des substances actives.
Conditions des essais.
Les essais contrôlés devraient correspondre aux bonnes pratiques agricoles limites proposées. Les conditions d'essai doivent prendre en considération les concentrations de résidus maximales pouvant raisonnablement se présenter (par exemple, nombre maximal d'applications proposées, utilisation de la dose maximale prévue, délais minimaux avant la récolte, périodes de rétention ou de stockage), mais restant représentatives des conditions réalistes les plus défavorables dans lesquelles la substance active serait utilisée.
Il faut produire et présenter un nombre suffisant de données pour confirmer que les schémas sont valables pour les régions et l'éventail des conditions susceptibles d'être rencontrées dans les régions en cause pour lesquelles l'utilisation du produit est recommandée.
Pour la fixation du programme d'essais contrôlés, il faudrait normalement prendre en considération des facteurs comme les différences climatiques entre zones de production, les différences dans les méthodes de production (par exemple, utilisation en plein air ou utilisation en serre), les époques de production, les types de formulation, etc.
En général, pour un ensemble comparable de conditions, les essais devraient être effectués sur au moins deux périodes de végétation. Toutes les dérogations devraient être pleinement justifiées.
Le nombre exact d'essais nécessaires peut difficilement être fixé sans évaluation préliminaire des résultats d'essai. Les exigences quant aux données minimales s'appliquent exclusivement si la comparabilité peut être établie entre zones de production, c'est-à-dire au sujet du climat, des méthodes et des périodes de végétation, etc. Dans l'hypothèse où toutes les autres variables (climat, etc.) sont comparables, un minimum de huit essais représentatifs de la zone de production proposée est exigé pour les grandes cultures. Pour les cultures d'importance mineure, il est normalement exigé quatre essais représentatifs de la zone de production proposée.
Etant donné le degré d'homogénéité intrinsèquement supérieur pour les résidus obtenus avec des traitements postérieurs à la récolte ou sur des cultures protégées, les essais relatifs à une période de production peuvent être acceptés. Pour les traitements postérieurs à la récolte, on exige, par principe, au moins quatre essais effectués de préférence en différents endroits sur diverses variétés. Une série d'essais doit être réalisée pour chaque méthode d'application et type de stockage, à moins qu'il soit possible d'identifier clairement la situation la plus défavorable quant aux résidus.
Le nombre d'études réalisées par période de végétation peut être réduit s'il peut être démontré que les niveaux de résidus dans les végétaux / produits végétaux seront inférieurs à la limite de détermination.
Si une partie significative de la culture comestible existe au moment de l'application, la moitié des essais contrôlés concernant les résidus relatés devrait inclure des données destinées à mettre en évidence l'effet du facteur temps sur la concentration de résidus présents (courbes de dégradation des résidus), sauf s'il peut être démontré que la culture comestible n'est pas touchée par l'application du produit phytopharmaceutique dans les conditions d'utilisation proposées.
6.4. Etudes sur l'alimentation des animaux.
But des essais.
L'objectif de ces études est de déterminer le taux de résidus contenus dans les produits animaux et provenant des résidus contenus dans les aliments pour animaux ou cultures fourragères.
Situations dans lesquelles les essais sont requis.
Les études relatives à l'alimentation des animaux ne sont requises que :
- si des concentrations significatives de résidus (0,1 mg/kg de la ration fourragère totale distribuée, sauf cas particuliers comme celui des substances actives qui s'accumulent) se produisent dans les végétaux ou parties de végétaux (par exemple résidus de nettoyage, déchets) utilisées pour l'alimentation animale ;
- si les études du métabolisme indiquent que des concentrations significatives de résidus (0,01 mg/kg ou une concentration supérieure à la limite de détermination, si celle-ci était supérieure à 0,01 mg/kg) peuvent se présenter dans tout tissu animal comestible, compte tenu des concentrations de résidus présentes dans les aliments potentiels pour animaux obtenues à la suite de l'administration d'une dose.
Le cas échéant, des études distinctes, relatives à l'alimentation des animaux pour les ruminants en lactation et/ou la volaille en période de ponte devraient être présentées. S'il ressort des études du métabolisme présentées conformément aux dispositions du point 6.2 que les voies métaboliques diffèrent dans des proportions significatives pour le porc comparativement aux ruminants, une étude sur l'alimentation des porcs doit être effectuée, sauf si l'on prévoit que l'absorption par les porcs ne sera pas significative.
Conditions des essais.
En général, l'aliment est administré selon trois dosages (concentration de résidus prévue, concentration trois à cinq fois supérieure et dix fois supérieure à la concentration prévue). La dose unique est calculée sur la base d'une ration fourragère théorique.
6.5. Effets de la transformation industrielle et/ou des préparations domestiques.
Situations dans lesquelles les essais sont requis.
La décision quant à la nécessité d'effectuer des études relatives à la transformation dépend :
- de la place prise par un produit transformé dans la ration alimentaire ou fourragère ;
- de la concentration des résidus dans le végétal ou produit végétal à transformer ;
- des propriétés physico-chimiques de la substance active ou des métabolites en cause et,
- de la possibilité que des produits de dégradation toxicologiquement significatifs puissent être découverts après la transformation des végétaux ou du produit végétal.
Des études relatives à la transformation ne sont normalement pas nécessaires si aucun résidu significatif ou décelable par analyse ne se présente dans le végétal ou le produit végétal à transformer, ou si la dose journalière théorique maximale est inférieure à 10 % de la dose journalière acceptable. En outre, des études relatives à la transformation ne sont normalement pas nécessaires pour les végétaux ou produits végétaux essentiellement consommés crus, à l'exception de ceux comprenant une fraction non comestible (par exemple agrumes, bananes, kiwis) où des données relatives à la distribution entre la peau et la pulpe peuvent être requises.
L'expression "résidu significatif" s'applique généralement à des concentrations supérieures à 0,1 mg/kg. Si le pesticide en cause se caractérise par une toxicité aiguë élevée et/ou une faible dose journalière acceptable, envisager d'effectuer des études relatives à la transformation pour des résidus décelables d'une concentration inférieure à 0,1 mg/kg.
Des études relatives aux effets sur la nature du résidu ne sont normalement pas requises si on n'applique normalement que des opérations physiques simples n'impliquant pas de changement de la température du végétal ou du produit végétal, comme le lavage, l'épluchage ou le pressage.
6.5.1. Effets sur la nature des résidus :
But des essais.
L'objectif des présentes études est de déterminer si la présence de résidus dans les produits crus entraîne ou non la formation de produits de dégradation ou de réaction pendant la transformation, ce qui peut nécessiter une évaluation séparée du risque.
Conditions de l'essai.
En fonction de la concentration et de la nature chimique du résidu contenu dans le produit cru, diverses conditions d'hydrolyse (simulant les opérations de transformation caractéristiques) représentatives doivent être examinées, le cas échéant. Il se peut aussi qu'il faille analyser les effets de processus autres que l'hydrolyse, lorsque les propriétés de la substance active ou des métabolites laissent supposer la présence de produits de dégradation toxicologiquement significatifs à la suite de ces processus. Les études sont normalement conduites avec une substance active radiomarquée.
6.5.2. Effets sur les concentrations de résidus :
But des essais.
Les principaux objectifs des présentes études sont les suivants :
- déterminer la distribution quantitative des résidus dans les divers produits intermédiaires et finis et estimer les facteurs de transfert :
- permettre une estimation plus réaliste de l'ingestion de résidus par la ration alimentaire ou fourragère.
Conditions des essais.
Les études relatives à la transformation doivent être représentatives des méthodes de transformation domestiques et/ou véritablement industrielles.
Dans le premier cas, il est habituellement nécessaire d'effectuer seulement un ensemble fondamental d'"études de bilan", représentatives des procédés communs applicables à des végétaux ou produits végétaux présentant des concentrations significatives de résidus. Le choix de ces procédés représentatifs doit être justifié. La technologie à appliquer dans les études relatives à la transformation devrait toujours correspondre, aussi étroitement que possible, aux conditions réelles normalement rencontrées. Il convient d'établir un bilan analysant sous l'angle de leur masse, les résidus contenus dans tous les produits intermédiaires et finis. L'établissement d'un tel bilan permet aussi de reconnaître toutes concentrations ou réductions des résidus dans les produits particuliers et de déterminer aussi les facteurs de transfert correspondants.
Si les produits végétaux transformés prennent une place importante dans la ration, et si l'"étude de bilan" indique qu'il pourrait se produire un transfert significatif de résidus dans le produit transformé, trois "études de suivi" doivent être effectuées en vue de déterminer la concentration des résidus ou les facteurs de dilution.
6.6. Résidus contenus dans les cultures suivantes.
But de l'essai.
L'objectif des présentes études est de permettre une évaluation des résidus pouvant être contenus dans les cultures suivantes.
Situations dans lesquelles les essais sont requis.
Si les données obtenues conformément à l'annexe I, point 7.1, ou à l'annexe II, point 9.1, démontrent que des concentrations significatives de résidus (supérieures à 10 % de la quantité de substance active appliquée représentant le total de la substance active non modifiée et de ses principaux métabolites ou produits de dégradation) demeurent dans le sol ou les produits végétaux, tels que la paille ou les matières organiques jusqu'à l'époque des semis ou de la plantation des cultures suivantes, et pourraient faire que la concentration de résidus soit supérieure à la limite de détermination dans les cultures suivantes au moment de la récolte, il convient d'examiner la situation quant aux résidus. Ceci comprend l'analyse de la nature du résidu contenu dans les cultures suivantes et suppose au moins une estimation théorique du niveau de ces résidus. Si la probabilité de présence de résidus dans les récoltes suivantes ne peut pas être exclue, des études du métabolisme et de la distribution devraient être effectuées et suivies, si nécessaire, d'essais en champs.
Conditions de l'essai.
Si une estimation théorique des résidus contenus dans les cultures suivantes est effectuée, donner des détails complets ainsi qu'une justification.
Si des études de métabolisme et de distribution ainsi que des essais en champs sont nécessaires, il convient de les effectuer sur des cultures représentatives choisies comme représentant une pratique agricole normale.
6.7. Limites maximales de résidus proposées et définition d'un résidu.
Les limites maximales de résidus proposées doivent être totalement justifiées notamment par la production, le cas échéant, des données complètes relatives à l'analyse statistique appliquée.
Pour juger des composés à inclure dans la définition d'un résidu, il convient de tenir compte de l'importance toxicologique des composés, des quantités pouvant être présentes et de l'applicabilité des méthodes d'analyse proposées pour le contrôle après autorisation et à des fins de suivi.
6.8. Propositions relatives aux délais d'attente avant récolte pour les utilisations envisagées, ou aux délais de rétention ou de stockage en cas d'utilisations postérieures à la récolte.
Les propositions doivent être entièrement justifiées.
6.9. Estimation de l'exposition potentielle ou réelle imputable au régime alimentaire ou à d'autres causes.
Il convient d'établir de manière réaliste la prévision de l'ingestion par le régime alimentaire ou fourrager, ce qui peut se faire de manière progressive et aboutir à une prévision de plus en plus réaliste de l'ingestion. Prendre éventuellement en considération d'autres sources d'exposition caractéristiques, tels que les résidus de médicaments, notamment vétérinaires.
6.10. Résumé et évaluation du comportement des résidus.
Un résumé et une évaluation de toutes les données exposées dans la présente section doivent être effectués conformément aux lignes directrices établies par les autorités compétentes des Etats membres au sujet du format de tels résumés et évaluations. Le document doit comprendre une estimation détaillée et critique de ces données dans le contexte des lignes directrices et critères importants pour l'évaluation et la prise de décision, une importance particulière étant accordée aux risques éventuels ou réels pour l'homme et les animaux et à l'importance, la qualité et la fiabilité de la base de données ainsi qu'à l'importance toxicologique de tout métabolite rencontré chez les animaux autres que les mammifères.
Un diagramme schématique doit être établi pour le trajet métabolique dans les végétaux et animaux avec une brève explication de la distribution et des modifications chimiques en cause.
7. Devenir et comportement dans l'environnement
(Arrêté du 1er février 1996, article 1er et Arrêté du 24 septembre 1996, article 5)
Introduction.
i) Les informations fournies jointes à celles concernant une ou plusieurs préparations contenant la substance active devront être suffisantes pour permettre une évaluation du devenir et du comportement de la substance active dans l'environnement ainsi que du comportement des espèces non cibles pouvant être menacées par une exposition à la substance active, ses métabolites et produits de dégradation et de réaction quand ils peuvent avoir une incidence toxicologique ou environnementale.
ii) En particulier, les informations fournies relatives à la substance active, jointes à d'autres informations pertinentes, ainsi que celles concernant une ou plusieurs préparations contenant la substance active devront être suffisantes pour :
- permettre une décision quant à l'inclusion éventuelle de la substance active dans l'annexe I ;
- fixer les conditions ou restrictions appropriées liées à toute inclusion sur la liste des substances actives ;
- classer la substance active quant aux risques ;
- fixer les symboles de danger, les indications relatives au danger et les phrases types relatives à la nature des risques et aux conseils de prudence pour la protection de l'environnement, à faire figurer sur l'emballage (conteneurs) ;
- prévoir la dispersion, le devenir et le comportement dans l'environnement de la substance active et des métabolites et produits de dégradation et de réaction significatifs ainsi que les durées correspondantes ;
- identifier les espèces et populations non cibles menacées en raison d'une exposition éventuelle,
et
- identifier les mesures nécessaires afin de minimiser la contamination de l'environnement et l'incidence sur les espèces non cibles.
iii) Une description détaillée (spécification) du matériau utilisé mentionnée à la section 1, point 11, doit être fournie. Lorsque les essais sont effectués avec la substance active, le matériau utilisé doit posséder les spécifications du produit utilisé dans la fabrication des préparations à autoriser, sauf s'il s'agit d'un produit radiomarqué.
Si des études sont effectuées avec de la substance active produite en laboratoire ou dans une installation pilote, elles doivent être répétées avec de la substance active fabriquée industriellement, sauf s'il peut être justifié que le matériau test utilisé est essentiellement le même pour les essais et les évaluations à caractère environnemental.
iv) Si les essais sont effectués à l'aide d'une substance radiomarquée, le marquage doit être situé à des emplacements (un ou plusieurs si nécessaire) permettant l'analyse des voies du métabolisme et de la dégradation ainsi que les études sur la dispersion de la substance active et de ses métabolites, produits de réaction et de dégradation dans l'environnement.
v) Il peut être nécessaire d'effectuer des études séparées concernant les métabolites, les produits de dégradation ou de réaction quand ces produits peuvent constituer un risque significatif pour les organismes non cibles ou la qualité de l'eau, du sol et de l'air et quand leurs effets ne peuvent être évalués à partir des résultats concernant la substance active. Avant d'effectuer ces études, il convient de tenir compte des informations des sections 5 et 6.
vi) Il convient, le cas échéant, de concevoir les essais et d'analyser les données sur la base de méthodes statistiques appropriées.
Les analyses statistiques doivent être décrites en détail (par exemple toutes les estimations ponctuelles doivent être fournies avec des intervalles de confiance, les valeurs de probabilité exactes plutôt que la mention significatif/non significatif).
7.1. Devenir et comportement dans le sol.
Toutes les informations pertinentes concernant le type et les propriétés du sol utilisé pour les études, y compris le pH, la teneur en carbone organique, la capacité d'échange cationique, la granulométrie et la capacité de rétention d'eau à pF = 0 et pF = 2,5 doivent être rapportées conformément aux normes ISO ou autres normes internationales applicables.
La biomasse microbienne des sols utilisés pour les études de dégradation en laboratoire doit être déterminée juste avant le début et à la fin de l'étude.
Il est recommandé d'utiliser, dans la mesure du possible, les mêmes sols au cours de toutes les études sur sol réalisées en laboratoire.
Les sols utilisés pour les études de dégradation ou de mobilité doivent être choisis en fonction de leur caractère représentatif de la gamme de sols typiques des différentes régions de la Communauté où l'utilisation existe ou est prévue et doivent :
- couvrir une gamme de teneur en carbone organique, de distribution granulométrique et de pH,
et
- couvrir les gammes de pH suivantes :
- 4,5 à 5,5 ;
- 6 à 7 et 8 (approximativement) quand, sur la base d'autres informations, il existe une suspicion de dégradation ou de mobilité dépendantes du pH (par exemple solubilité et taux d'hydrolyse "points 2.7 et 2.8").
Les sols utilisés doivent, dans la mesure du possible, être fraîchement prélevés. Si l'utilisation de sols stockés est inévitable, le stockage doit être effectué de manière adéquate pendant une durée limitée, dans des conditions définies et rapportées. Les sols stockés pendant des périodes plus longues ne peuvent être utilisés que pour des études d'absorption et de désorption.
Le sol sélectionné pour effectuer les études ne devra pas présenter de caractéristiques extrêmes en ce qui concerne des paramètres tels que la distribution granulométrique, la teneur en carbone organique et le pH.
Les sols devront être prélevés et manipulés conformément aux normes ISO 10381-6 (qualité des sols - échantillonnage - guide du prélèvement, de la manipulation et du stockage des sols pour l'évaluation des processus microbiens en laboratoire). Tous les écarts doivent être rapportés et justifiés.
Les études au champ doivent être effectuées dans des conditions aussi proches que possible de la pratique agricole normale sur une gamme de types de sol et de conditions climatiques représentative de la (des) zone(s) d'utilisation.
Les conditions météorologiques doivent être indiquées dans le cas d'études au champ.
7.1.1. Voie et vitesse de dégradation.
7.1.1.1. Voie de dégradation.
But des essais.
Les données et informations fournies, jointes à d'autres données et informations pertinentes, devront être suffisantes pour permettre :
- d'identifier, dans la mesure du possible, l'importance relative des types de processus mis en jeu (importance relative de la dégradation chimique et de la dégradation biologique) ;
- d'identifier les composés présents représentant constamment plus de 10 p. 100 de la quantité de substance active ajoutée ainsi que, dans la mesure du possible, les résidus non extractibles ;
- d'identifier également, dans la mesure du possible, les composés représentant moins de 10 p. 100 de la quantité de substance active ajoutée ;
- d'établir les proportions relatives des composés (bilan massique),
et
- de définir le résidu dans le sol auquel les espèces non cibles sont exposées ou peuvent l'être.
Lorsqu'il est fait référence aux résidus non extractibles, ceux-ci sont définis comme des espèces chimiques, provenant des pesticides utilisés conformément aux bonnes pratiques agricoles, ne pouvant être extraites à l'aide de méthodes qui ne modifient pas sensiblement la nature chimique de ces résidus. Ces résidus non extractibles ne sont pas supposés inclure des fragments formés par des voies métaboliques conduisant à des produits naturels.
7.1.1.1.1. Dégradation aérobie.
Situation dans lesquelles les essais sont requis.
La (les) voie(s) de dégradation doit (doivent) toujours être décrite(s) sauf quand la nature et le mode d'utilisation des préparations contenant la substance active excluent la possibilité d'une contamination du sol comme dans le cas des emplois sur des produits stockés ou des traitements de cicatrisation pour les arbres.
Modalités des essais.
La (les) voie(s) de dégradation doit (doivent) être décrite(s) pour un seul sol.
Les résultats obtenus doivent être présentés sous forme de schémas où figurent les voies concernées et sous forme de bilan présentant la distribution du marquage radioactif en fonction du temps entre :
- la substance active ;
- le CO2 ;
- les composants volatils autres que le CO2 ;
- les produits de transformation individuels identifiés ;
- les substances extractibles non identifiées,
et
- les résidus non extractibles présents dans le sol.
L'étude des voies de dégradation doit comprendre toutes les opérations possibles permettant de caractériser et de quantifier les résidus non extractibles formés au bout d'une période de 100 jours quand ils dépassent 70 p. 100 de la substance active appliquée. Le choix des meilleures techniques et méthodologies à appliquer s'effectue cas par cas. Une justification doit être fournie quand les composants impliqués ne sont pas caractérisés.
La durée normale de l'étude est de 120 jours, sauf quand, au bout d'une période plus courte, les taux de résidus non extractibles et de CO2 peuvent être extrapolés avec certitude à une période de 100 jours.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides Society of Environmental Toxicology and Chemistry (Setac), 1995. Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides, ISBN 90-5607-002-9.
7.1.1.1.2. Etudes complémentaires.
Dégradation anaérobie.
Situations dans lesquelles les essais sont requis.
Une étude de dégradation anaérobie doit être rapportée à moins qu'il puisse être justifié que l'exposition à des conditions anaérobies des produits phytopharmaceutiques concernant la substance active n'est pas probable.
Modalités et ligne directrice des essais.
Mêmes dispositions que celles des titres correspondants au point 7.1.1.1.1.
Photodégradation dans le sol.
Situations dans lesquelles les essais sont requis.
Une étude de photodégradation dans le sol doit être rapportée à moins qu'il ne puisse être justifié que le dépôt de la substance active à la surface du sol n'est pas probable.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
7.1.1.2. Vitesse de dégradation.
7.1.1.2.1. Etudes de laboratoire.
But des essais.
Les études de dégradation dans le sol devront fournir les meilleures estimations possibles du temps nécessaire à la dégradation de 50 p. 100 et de 90 p. 100 (DT50 lab et DT90 lab) de la substance active ainsi que des métabolites et produits de réaction et de dégradation ayant une incidence toxicologique et environnementale dans des conditions de laboratoire.
Dégradation aérobie.
Situations dans lesquelles les essais sont requis.
La vitesse de dégradation dans le sol doit toujours être rapportée, sauf quand la nature et le mode d'utilisation des produits phytopharmaceutiques contenant la substance active excluent la possibilité d'une contamination du sol comme c'est le cas pour les emplois sur des produits stockés ou les traitements de cicatrisation des arbres.
Modalités des essais.
La vitesse de dégradation aérobie de la substance active dans trois types de sol en plus des informations citées au point 7.1.1.1.1 doit être rapportée.
Une étude supplémentaire doit être effectuée à 10° C sur l'un des sols utilisés pour l'étude de la dégradation à 20° C afin d'étudier l'incidence de la température sur la dégradation jusqu'à ce que l'on dispose d'un modèle validé de calcul communautaire pour l'extrapolation des vitesses de dégradation aux basses températures.
La durée normale de l'étude est de 120 jours sauf si plus de 90 p. 100 de la substance active sont dégradés avant l'expiration de cette période.
Des études similaires pour trois types de sols doivent être rapportées pour tous les métabolites et produits de dégradation et de réaction qui sont présents dans les sols et qui représentent à tout moment de l'étude plus de 10 p. 100 de la quantité de substance active ajoutée, sauf quand il est possible de calculer leurs valeurs de DT50 à partir des résultats des études de dégradation réalisées avec la substance active.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
Dégradation anaérobie.
Situations dans lesquelles les essais sont requis.
La vitesse de dégradation en conditions anaérobies de la substance active doit être étudiée quand une étude anaérobie doit être effectuée conformément au point 7.1.1.1.2.
Modalités des essais.
La vitesse de dégradation en conditions anaérobies de la substance active doit être étudiée dans le sol utilisé pour l'étude anaérobie effectuée conformément au point 7.1.1.1.2.
La durée normale de l'étude est de 120 jours sauf si plus de 90 p. 100 de la substance active sont dégradés avant l'expiration de cette période.
Des études similaires pour un type de sol doivent être relatées pour tous les métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale qui sont présents dans le sol et représentent à tout moment de l'étude plus de 10 p. 100 de la quantité de substance active ajoutée, sauf quand il est possible de calculer leurs valeurs de DT50 à partir des résultats des études de dégradation réalisées avec la substance active.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
7.1.1.2.2. Etudes au champ.
Etudes de dissipation dans le sol.
But des essais.
Les études de dissipation dans le sol devraient fournir des estimations du temps nécessaire à la dissipation de 50 p. 100 et de 90 p. 100 (DT50f et DT90f) de la substance active dans des conditions de terrain. Si nécessaire, des informations concernant les métabolites et les produits de dégradation et de réaction ayant une incidence toxicologique et environnementale doivent être mentionnées.
Situations dans lesquelles les essais sont requis.
Les essais doivent être effectués quand la DT50 lab déterminée à une température de 20° C et avec une teneur en humidité du sol correspondant à une proche d'une valeur de pF de 2 à 2,5 (succion) est supérieure à 60 jours.
Si les produits phytopharmaceutiques contenant la substance active sont destinés à une utilisation dans des conditions climatiques froides, les essais doivent être effectués quand la DT 50 lab, déterminée à une température de 10° C et avec une teneur en humidité du sol correspondant à une valeur de pF de 2 à 2,5 (succion) est supérieure à quatre-vingt-dix jours.
Modalités des essais.
Les études individuelles effectuées sur une gamme de sols représentatifs (en général quatre types de sol différents) doivent être poursuivies jusqu'à ce que plus de 90 p. 100 de la quantité utilisée se soient dissipés. La durée maximale de ces études est de 24 mois.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
Etudes de résidus dans le sol.
But des essais.
Les études de résidus dans le sol doivent fournir des estimations des niveaux de résidus dans le sol à la récolte ou au moment du semis ou de la mise en place des cultures suivantes.
Situations dans lesquelles les essais sont requis.
Les études de résidus dans le sol doivent être rapportées quand la DT 50 lab est supérieure à un tiers de la période qui va de l'application à la récolte et quand l'absorption par la culture suivante est possible, sauf quand les résidus dans le sol au moment du semis ou de la mise en place de la culture suivante peuvent être évalués avec fiabilité à partir des données fournies par les études de dissipation dans le sol ou quand il peut être justifié que ces résidus ne peuvent pas être phytotoxiques ou atteindre un niveau de résidus inacceptable dans les rotations culturales.
Modalités des essais.
Les études doivent être poursuivies jusqu'à la récolte ou jusqu'au semis ou la mise en place des cultures suivantes, à moins que plus de 90 p. 100 de la quantité appliquée ne se soient dissipés.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
Etudes d'accumulation dans le sol.
But des essais.
Les essais devront fournir des données suffisantes permettant d'évaluer la possibilité d'accumulation de résidus de la substance active, des métabolites et des produits de dégradation et de réaction ayant une incidence toxicologique et environnementale.
Situations dans lesquelles les essais sont requis.
Quand, sur la base des études de dissipation dans le sol, il est établi que la DT 90f est supérieure à un an et quand une application répétée est envisagée, au cours de la même période de végétation ou d'années successives, la possibilité d'accumulation de résidus dans le sol et le niveau auquel une concentration plateau est atteinte doivent être étudiés sauf quand des informations fiables peuvent être fournies par calcul au moyen d'un modèle ou un autre type d'évaluation approprié.
Modalités des essais.
Les études au champ de longue durée doivent être effectuées sur deux sols appropriés et comporter des applications multiples.
Avant d'effectuer ces études, le demandeur doit obtenir l'accord des autorités compétentes sur le type d'étude à effectuer.
7.1.2. Adsorption et désorption.
But des essais.
Les données et informations fournies, jointes à d'autres données et informations pertinentes, devront être suffisantes pour déterminer le coefficient d'adsorption de la substance active et des métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale.
Situations dans lesquelles les essais sont requis.
Les études doivent toujours être rapportées sauf quand la nature et le mode d'utilisation des préparations contenant la substance active excluent toute possibilité de contamination du sol comme dans le cas des emplois sur des produits stockés ou des traitements de cicatrisation pour les arbres.
Modalités des essais.
Les études sur la substance active doivent être rapportées pour quatre types de sol.
Pour au moins trois types de sol, des études similaires doivent être rapportées pour tous les métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale et qui, dans les études de dégradation dans le sol, représentent à tout moment plus de 10 p. 100 de la quantité de substance active ajoutée.
Ligne directrice. Méthode OCDE 106.
7.1.3. Mobilité dans le sol.
7.1.3.1. Etudes de lixiviation sur colonne.
But des essais.
L'essai doit fournir des données suffisantes pour évaluer la mobilité et le potentiel de lixiviation de la substance active et, si possible, des métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale.
Situations dans lesquelles les essais sont requis.
Les études portant sur quatre sols doivent être effectuées quand il n'est pas possible d'obtenir des valeurs fiables des coefficients d'adsorption dans les études d'adsorption et de désorption fournies par l'application du point 7.1.2.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
7.1.3.2. Lixiviation sur colonne de résidus vieillis.
But des essais.
Les essais doivent fournir des données suffisantes pour estimer la mobilité et le potentiel de lixiviation des métabolites et des produits de dégradation et de réaction ayant une incidence environnementale.
Situations dans lesquelles les essais sont requis.
Les études doivent être effectuées sauf :
- quand la nature et le mode d'utilisation des préparations contenant la substance active excluent toute possibilité de contamination du sol comme dans le cas des emplois sur des produits stockés ou des traitements de cicatrisation des arbres,
ou
- quand une étude distincte relative aux métabolites et aux produits de dégradation ou de réaction a été effectuée conformément au point 7.1.2 ou au point 7.1.3.1.
Modalités des essais.
La (les) période(s) de vieillissement doit(vent) être déterminée(s) sur la base d'un examen du schéma de dégradation de la substance active et des métabolites afin de garantir la présence d'un spectre pertinent de métabolites au moment de la lixiviation.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
7.1.3.3. Etudes lysimétriques ou études de lixiviation au champ.
But des essais.
Les essais doivent fournir des données concernant :
- la mobilité dans le sol ;
- le potentiel de lixiviation vers les eaux souterraines ;
- la dispersion potentielle dans le sol.
Situations dans lesquelles les essais sont requis.
L'avis des spécialistes sera nécessaire afin de décider si les études lysimétriques ou les études de lixiviation au champ doivent être effectuées compte tenu des résultats des études de dégradation et d'autres études de mobilité et des concentrations environnementales prévisibles dans les eaux souterraines (PECGW) calculées conformément aux dispositions de l'annexe II, section 9. Le type et les conditions de l'étude à mener doivent faire l'objet d'une discussion avec les autorités compétentes.
Modalités des essais.
La conception des dispositifs expérimentaux et des études individuelles requiert le plus grand soin afin de garantir que les résultats obtenus puissent être utilisés à des fins d'évaluation. Les études doivent inclure la situation du cas réaliste le plus défavorable, compte tenu du type de sol, des conditions climatiques, de la dose d'application, et de la fréquence et de la période d'application.
Il convient d'analyser l'eau percolée au travers des colonnes de sol à intervalles appropriés et de déterminer les résidus dans les végétaux à la récolte. Les résidus contenus dans au moins cinq couches du profil de sol doivent être déterminés en fin d'essai. Il convient d'éviter les prélèvements intermédiaires d'échantillons, étant donné que l'extraction de végétaux (sauf pour la récolte conformément à la pratique agricole normale) et de carottes de sol influence le processus de lixiviation.
Il convient d'effectuer des relevés de précipitations, de température du sol et de l'air à intervalles réguliers (au moins une fois par semaine).
Etudes lysimétriques.
Modalités des essais.
La profondeur minimale des lysimètres doit être de 100 cm. La profondeur maximale doit être de 130 cm. Les monolithes de sol doivent être intactes. Les températures du sol doivent être similaires aux température du terrain, une irrigation supplémentaire doit éventuellement être pratiquée afin de garantir une croissance optimale des végétaux et une infiltration de l'eau en quantité similaire à celle des régions pour lesquelles une autorisation est demandée. Quand, au cours de l'étude, le sol doit être remanié pour des raisons agricoles, il ne doit pas l'être sur une profondeur dépassant 25 cm.
Etudes de lixiviation du champ.
Modalités des essais.
Des informations relatives au niveau piézométrique de la nappe phréatique dans les champs d'essais doivent être fournies. Si des fissurations du sol sont observées au cours de l'étude, elles doivent faire l'objet d'une description exhaustive.
Une attention particulière doit être portée au nombre et à l'emplacement des dispositifs de prélèvement de l'eau. L'installation dans le sol de ces dispositifs ne doit pas donner lieu à l'apparition de voies d'infiltration privilégiées.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
7.2. Devenir et comportement dans l'eau et l'air.
But des essais.
Les informations et données fournies, jointes à celles concernant une ou plusieurs préparations contenant la substance active, ainsi que d'autres informations pertinentes devront être suffisantes pour permettre d'établir ou d'estimer :
- la persistance dans les systèmes aquatiques (sédiments de fond et eau, y compris les matières en suspension) ;
- le niveau de danger auquel les organismes vivant dans les sédiments, l'eau et l'air sont exposés ;
- le potentiel de contamination des eaux de surface et des eaux souterraines.
7.2.1. Voie et vitesse de dégradation dans les systèmes aquatiques (autres aspects qu'au point 2.9).
But des essais.
Les données et les informations fournies, jointes à d'autres données et informations pertinentes, devront être suffisantes pour :
- identifier l'importance relative des types de processus impliqués (importance relative de la dégradation chimique et de la dégradation biologique) ;
- identifier, dans la mesure du possible, les différents composés ;
- établir les proportions relatives des composés présents et leur distribution entre l'eau, matières en suspension incluses, et le sédiment,
et
- permettre de définir les résidus auxquels les espèces non cibles sont exposées ou peuvent l'être.
7.2.1.1. Hydrolyse.
Situations dans lesquelles les essais sont requis.
Il convient toujours d'effectuer des essais ayant pour objet les métabolites et les produits de dégradation et de réaction ayant une incidence toxicologique et environnementale et représentant à tout moment plus de 10 p. 100 de la quantité de substance active ajoutée sauf si l'on dispose d'informations suffisantes relatives à leur dégradation grâce au test effectué conformément au point 2.9.1.
Modalités et ligne directrice des essais.
Mêmes dispositions qu'aux titres correspondants du point 2.9.1.
7.2.1.2. Dégradation photochimique.
Situations dans lesquelles les essais sont requis.
Les essais ayant pour objet les métabolites et les produits de dégradation et de réaction ayant une incidence toxicologique et environnementale et représentant à tout moment plus de 10 p. 100 de la quantité de substance active ajoutée doivent toujours être réalisés sauf si l'on dispose d'informations suffisantes relatives à leur dégradation grâce au test effectué conformément aux points 2.9.2 et 2.9.3.
Modalités des essais et ligne directrice.
Mêmes dispositions qu'aux titres correspondants des points 2.9.2 et 2.9.3.
7.2.1.3. Dégradation biologique.
7.2.1.3.1. Biodégradabilité facile.
Situations dans lesquelles les essais sont requis.
L'essai doit toujours être réalisé sauf s'il n'est pas exigé conformément aux dispositions de l'annexe VI de la directive 67/548/CEE concernant la classification de la substance active.
Ligne directrice. Méthode C4 CEE.
7.2.1.3.2. Etude de système eau-sédiment.
Situations dans lesquelles les essais sont requis.
L'essai doit toujours être rapporté sauf s'il peut être justifié qu'aucune contamination des eaux de surface n'est possible.
Ligne directrice.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
7.2.1.4. Dégradation dans la zone saturée.
Situations dans lesquelles les essais sont requis.
Les taux de transformation, dans la zone saturée, des substances actives et des métabolites, des produits de dégradation et de réaction ayant une incidence toxicologique et environnementale peuvent fournir des informations utiles sur le devenir de ces substances dans les eaux souterraines.
Modalités des essais.
L'avis des spécialistes est requis pour déterminer si ces informations sont nécessaires. Avant d'effectuer ces études, le pétitionnaire doit obtenir l'accord des autorités compétentes sur le type d'étude à effectuer.
7.2.2. Voie et vitesse de dégradation dans l'air (autres aspects qu'au point 2.10).
Instructions en cours d'élaboration.
7.3. Définition du résidu.
En fonction de la composition chimique des résidus présents dans le sol, l'eau ou l'air résultant de l'utilisation ou de l'utilisation proposée d'un produit phytopharmaceutique contenant la substance active, il convient de faire une proposition pour la définition du résidu en tenant compte à la fois des niveaux relevés et de leur incidence toxicologique et environnementale.
7.4. Données de surveillance.
Les données de surveillance disponibles concernant le devenir et le comportement de la substance active et des métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale doivent être rapportées.
8. Etudes écotoxicologiques
(Arrêté du 1er février 1996, article 1er et Arrêté du 24 septembre 1996, article 5)
Introduction.
i) Les informations fournies, jointes à celles qui concernent une ou plusieurs préparations contenant la substance active, doivent être suffisantes pour permettre d'évaluer l'impact sur les espèces non cibles (flore et faune) dont l'exposition à la substance active, à ses métabolites et à ses produits de dégradation et de réaction peut être dangereuse, lorsque ces produits ont une importance pour l'environnement. L'impact peut être dû à une exposition unique et prolongée et peut être réversible ou irréversible.
ii) En particulier, les informations fournies sur la substance active, jointes aux autres informations pertinentes et aux informations relatives à une ou plusieurs préparations contenant cette substance active, doivent être suffisantes pour :
- décider si la substance active peut ou non être incluse dans la liste des substances actives ;
- spécifier les conditions ou restrictions appropriées devant accompagner toute inclusion dans la liste des substances actives ;
- permettre une évaluation des risques à court aussi bien qu'à long terme pour les espèces non cibles (populations, communautés et processus, selon le cas) ;
- classer la substance active en fonction du danger ;
- préciser les précautions à prendre pour protéger les espèces non cibles ;
- définir les symboles des dangers, les indications du danger et les phrases relatives à la nature des risques ainsi qu'aux conseils de prudence pour la protection de l'environnement à faire figurer sur les emballages (conteneurs).
iii) Il est nécessaire de faire état de tous les effets potentiellement néfastes constatés au cours des investissements écotoxicologiques de routine et de réaliser et rapporter, si les autorités compétentes le demandent, les études supplémentaires qui se révéleraient nécessaires pour déceler les éventuels mécanismes en cause et évaluer la signification de ces effets. Il est indispensable de rapporter toutes les données et les informations biologiques disponibles concourant à l'évaluation du profil écotoxicologique de la substance active.
iv) Les informations sur le sort et le comportement dans l'environnement, établies et présentées conformément aux points 7.1 à 7.4 et sur les niveaux des résidus dans les végétaux établis et présentés conformément au chapitre 6, sont essentielles pour l'évaluation de l'impact sur les espèces non cibles, car, associées aux renseignements sur la nature de la préparation et son mode d'emploi, elles définissent la nature et l'ampleur de l'exposition potentielle. Les études et les informations toxicocinétiques et toxicologiques présentées conformément aux points 5.1 et 5.8 fournissent des données essentielles sur la toxicité pour les vertébrés et les mécanismes en cause.
v) S'il y a lieu, des essais devraient être mis au point et les données obtenues devraient être analysées à l'aide de méthodes statistiques appropriées. Tous les détails de l'analyse statistique sont à noter (par exemple, toutes les estimations doivent être délimitées par un intervalle de confiance, il vaut mieux indiquer des valeurs "p" exactes au lieu de préciser qu'une valeur est significative/non significative).
Substance d'essai.
vi) Il est indispensable de fournir une description détaillée (spécification) du produit utilisé, conformément aux dispositions du point 1.11. Lorsque les essais sont effectués au moyen de la substance active, le produit utilisé devrait répondre aux spécifications qui seront retenues dans la fabrication des préparations à autoriser, sauf en cas d'utilisation de produit marqué.
vii) Lorsque les études sont réalisées sur la substance active produite en laboratoire ou dans une unité de production pilote, elles devront être répétées sur la substance active fabriquée, à moins qu'il puisse être prouvé que la substance d'essai utilisée est essentiellement la même aux fins des essais et de l'évaluation du comportement écotoxicologique. En cas de doute, il y a lieu de présenter des études de recoupement appropriées permettant de trancher quant à l'éventuelle nécessité de répéter les études.
viii) Dans le cas d'études prévoyant une administration prolongée pendant une certaine période, cette administration devrait être effectuée, de préférence, au moyen d'un seul lot de substance active, si la stabilité de celle-ci le permet.
Si une étude comporte l'utilisation de doses différentes, la relation entre dose et effet néfaste doit être notée.
ix) Dans toutes les études d'alimentation, il y a lieu d'indiquer la dose moyenne administrée, voire, si possible, la dose en mg/kg de poids corporel. Lorsque l'administration s'effectue par les rations alimentaires, le composé d'essai doit y être distribué uniformément.
x) Des études séparées sur les métabolites, les produits de dégradation ou de réaction peuvent s'imposer lorsque ces composés peuvent présenter un risque non négligeable pour les organismes non cibles et que les résultats des études portant sur la substance active ne permettent pas d'évaluer leurs effets. Avant de procéder à ces études, il convient de tenir compte des informations résultant des chapitres 5, 6 et 7.
Organismes d'essai.
xi) Pour faciliter l'évaluation de la signification des résultats obtenus, y compris l'estimation de la toxicité intrinsèque et des facteurs influençant la toxicité, il faudrait utiliser, dans la mesure du possible, des individus appartenant à la même souche ou à la même origine certifiée de l'espèce faisant l'objet des différents essais de toxicité.
8.1 Effets sur les oiseaux.
8.1.1. Toxicité orale aiguë.
Objet de l'essai.
Dans la mesure du possible, l'essai devrait permettre d'établir les valeurs de la DL 50, la dose seuil létale, les temps de réponse et de récupération et le NSEO et doit faire état des observations pathologiques significatives à l'autopsie.
Circonstances où l'essai est requis.
Il est indispensable de rechercher les effets éventuels de la substance active sur les oiseaux, sauf dans les cas où la substance active est destinée exclusivement à l'incorporation dans des préparations à utiliser uniquement dans des espaces clos (tels que des serres ou des entrepôts de denrées alimentaires).
Conditions de l'essai.
La toxicité orale aiguë de la substance active pour une espèce de caille (japonaise) (Coturnix coturnix japonica) ou colin de Virginie (Colinus virginianus) ou pour le canard colvert (Anas platyrhynchos) doit être déterminée. La dose maximale utilisée dans les essais ne dépassera pas 2 000 mg/kg de poids corporel.
Lignes directrices.
Setac. Procédures d'évaluation du comportement environnemental et de l'écotoxicité des pesticidesSociety of Environmental Toxicology and Chemistry (Setac), 1995. Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides, ISBN 90-5607-002-9.
8.1.2. Toxicité alimentaire à court terme.
Objet de l'essai.
L'essai devrait permettre d'établir la toxicité alimentaire à court terme (la CL 50, la plus faible concentration létale, si possible la concentration sans effet observé - CSEO -, les temps de réponse et de récupération) et faire état des observations pathologiques significatives à l'autopsie.
Circonstances où l'essai est requis.
La toxicité alimentaire (cinq jours) de la substance active pour les oiseaux doit toujours être testée sur une espèce, sauf lorsqu'il est fait état d'une étude réalisée selon les dispositions du point 8.1.3. Lorsque le NSEO oral aigu est m 500 mg/kg de poids corporel ou lorsque la CSEO à court terme est l 500 mg/kg d'aliment, l'essai doit être répété sur une deuxième espèce.
Conditions de l'essai.
La première espèce étudiée doit être la caille ou le canard colvert. Si une deuxième espèce doit être testée, elle ne doit pas être apparentée à la première.
Lignes directrices.
L'essai doit être réalisé en conformité avec la méthode 205 de l'OCDE.
8.1.3. Toxicité subchronique et reproduction.
Objet de l'essai.
L'essai devrait permettre d'établir la toxicité subchronique de la substance active pour les oiseaux ainsi que la toxicité pour leur reproduction.
Circonstances où l'essai est requis.
Il est obligatoire de déterminer la toxicité subchronique ou de reproduction de la substance active pour les oiseaux, sauf s'il est prouvé que l'exposition continue ou répétée des adultes ou l'exposition des sites de nidification pendant la période de reproduction est improbable.
Lignes directrices.
L'essai doit être réalisé en conformité avec la méthode 206 de l'OCDE.
8.2. Effets sur les organismes aquatiques.
Les données des essais visés aux points 8.2.1, 8.2.4 et 8.2.6 doivent être soumises pour toutes les substances actives, même s'il n'est pas attendu que les produits phytopharmaceutiques qui la contiennent puissent atteindre les eaux de surface dans les conditions d'emploi proposées. Ces données sont requises en vertu des dispositions de l'annexe VI de la directive 67/548/CEE pour le classement de la substance active.
Les données rapportées doivent être étayées par des données analytiques sur les concentrations de la substance d'essai dans les milieux d'essai.
8.2.1. Toxicité aiguë pour les poissons.
Objet de l'essai.
L'essai devrait permettre d'établir la toxicité aiguë (CL 50) et exposer les détails des effets observés.
Circonstances où l'essai est requis.
L'essai doit toujours être réalisé.
Conditions de l'essai.
La toxicité aiguë de la substance active doit être déterminée pour la truite arc-en-ciel (Oncorhynchus mykiss) et pour une espèce de poisson des eaux chaudes. Lorsque des essais doivent être effectués sur les métabolites, les produits de dégradation ou de réaction, l'espèce retenue sera la plus sensible des deux espèces dans l'essai d'exposition à la substance active.
Lignes directrices.
L'essai doit être effectué conformément à l'annexe de la directive 92/69/CEE de la Commission portant dix-septième adaptation au progrès technique de la directive 67/548/CEE concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives au classement, à l'emballage et à l'étiquetage des substances dangereuses, méthode C 1.
8.2.2. Toxicité chronique pour les poissons.
Circonstances où l'essai est requis.
Une étude de toxicité chronique doit toujours être effectuée, sauf s'il est prouvé que l'exposition continue ou répétée des poissons est improbable ou que l'on dispose d'une étude adéquate de microcosme ou de mésocosme.
Un jugement d'expert est nécessaire pour décider des essais à effectuer. En particulier, lorsqu'il s'agit d'une substance active à risques (liés à sa toxicité pour le poisson ou à son exposition potentielle), le demandeur doit soumettre le type d'essai à effectuer à l'agrément des autorités compétentes.
Un essai de toxicité pour les poissons dans les premières phases de la vie peut être approprié lorsque le FBC est compris entre 100 et 1 000 ou que le CL 50 de la substance active est < 0,1 mg/l.
Un essai du cycle biologique des poissons peut être approprié :
- lorsque le facteur de bioconcentration est supérieur à 1 000 et que l'élimination de la substance active au cours d'une phase d'épuration de 14 jours est inférieure à 95 p. 100,
ou
- que la substance est stable dans l'eau ou dans le sédiment (DT 90 > 100 jours).
Les poissons juvéniles ne doivent pas faire l'objet d'un essai de toxicité chronique lorsqu'un essai de toxicité sur les premières phases de la vie ou qu'un essai sur le cycle biologique a été effectué de même, il n'est pas nécessaire de procéder à un essai de toxicité sur les premières phases de la vie lorsqu'un essai sur le cycle biologique a été effectué.
8.2.2.1. Essai de toxicité chronique pour les poissons juvéniles.
Objet de l'essai.
L'essai devrait permettre d'établir les effets sur la croissance, le niveau seuil des effets létaux ou des effets observés, la CSEO et les détails des effets observés.
Conditions de l'essai.
L'essai doit être pratiqué sur la truite arc-en-ciel juvénile, après une exposition de 28 jours à la substance active. Il doit fournir des informations sur les effets sur la croissance et le comportement.
8.2.2.2. Essai de toxicité pour les poissons dans les premières phases de la vie.
Objet de l'essai.
L'essai devrait permettre d'établir les effets sur la croissance, le niveau seuil des effets létaux et des effets observés, la CSEO et les détails des effets observés.
Directives.
L'essai doit être effectué en conformité avec la méthode 210 de l'OCDE.
8.2.2.3. Essai sur le cycle biologique des poissons.
Objet de l'essai.
L'essai permettra d'établir les effets sur la reproduction des générations parentales et sur la viabilité des générations de descendants.
Conditions de l'essai.
Le demandeur doit solliciter au préalable l'agrément des autorités compétentes sur la nature et les conditions de l'essai à effectuer.
8.2.3. Bioconcentration chez le poisson.
Objet de l'essai.
L'essai devrait permettre d'établir les facteurs de la bioconcentration à l'équilibre (FBC), les constantes des taux d'accumulation et des taux d'élimination, calculés pour chaque substance d'essai, ainsi que les limites de confiance correspondantes.
Circonstances où l'essai est requis.
Le potentiel de bioconcentration des substances actives, des métabolites et des produits de dégradation ou de réaction, susceptibles de se déposer dans les tissus adipeux (tel que le log POE 3 - voir chapitre 2, point 2.8, ou d'autres indications relatives à la bioconcentration), doit être déterminé et rapporté, à moins qu'il ne soit prouvé qu'une exposition entraînant une bioconcentration est improbable.
Lignes directrices.
L'essai doit être effectué en conformité avec la méthode 305 E de l'OCDE.
8.2.4. Toxicité aiguë pour les invertébrés aquatiques.
Objet de l'essai.
L'essai devrait permettre d'établir la toxicité aiguë à 24 et 48 heures de la substance active, exprimée sous forme de concentration médiane effective (CE 50) provoquant l'immobilisation et, si possible, la concentration la plus élevée ne provoquant pas d'immobilisation.
Circonstances où l'essai est requis.
La toxicité aiguë doit toujours être déterminée pour Daphnia (de préférence, Daphnia magna). Lorsque des produits phytopharmaceutiques contenant la substance active doivent être utilisés directement dans ou sur des eaux de surface, des données additionnelles doivent être communiquées au sujet d'au moins une espèce représentative de chacun des groupes suivants : insectes aquatiques, crustacés aquatiques (d'une espèce non apparentée à Daphnia) et mollusques aquatiques.
Lignes directrices.
L'essai doit être effectué en conformité avec la directive 92/69/CEE, méthode C 2.
8.2.5. Toxicité chronique pour les invertébrés aquatiques.
Objet de l'essai.
L'essai devrait permettre d'établir, si possible, les valeurs de la CE 50 concernant des effets tels que l'immobilisation et la reproduction ainsi que la concentration la plus élevée n'ayant aucun effet sur la mortalité ou la reproduction (CSEO) et des détails des effets observés.
Circonstances où l'essai est requis.
L'essai sur Daphnia et sur au moins une espèce représentative d'insecte aquatique et une espèce de mollusque gastéropode aquatique est obligatoire à moins qu'il ne soit prouvé qu'une exposition continue ou répétée est improbable.
Conditions de l'essai.
L'essai sur Daphnia doit avoir une durée de 21 jours.
Lignes directrices.
L'essai doit être effectué en conformité avec la méthode 202 partie II de l'OCDE.
8.2.6. Effets sur la croissance des algues.
Objet de l'essai.
L'essai devrait permettre d'établir les valeurs des CE 50 relatives à la croissance et au taux de croissance, les valeurs de la CSEO et les détails des effets observés.
Circonstances où l'essai est requis.
Les effets éventuels de la substance active sur la croissance des algues doivent toujours être consignés.
Pour les herbicides, l'essai doit être pratiqué sur une seconde espèce d'un autre groupe taxonomique.
Lignes directrices.
L'essai doit être effectué en conformité avec la directive 92/69/CEE, méthode C 3.
8.2.7. Effets sur les organismes vivant dans les sédiments.
Objet de l'essai.
L'essai permettra de mesurer les effets sur la survie et le développement (y compris les effets sur la sortie des adultes de l'espèce Chironomus), les valeurs des CE 50 correspondantes et les valeurs des CSEO.
Circonstances où l'essai est requis.
Lorsque les données sur le sort et le comportement dans l'environnement prescrites au chapitre 7 indiquent qu'une substance active risque de se répartir et de persister dans les sédiments aquatiques, un jugement d'expert est nécessaire pour décider s'il faut réaliser un essai de toxicité aiguë ou chronique pour les sédiments. L'avis susmentionné devra établir la probabilité des effets sur les invertébrés vivant dans les sédiments par une comparaison des valeurs des CE 50 relatives à la toxicité pour les invertébrés aquatiques visées aux points 8.2.4 et 8.2.5 avec les niveaux prédits de la substance active dans les sédiments tirés des données visées au chapitre 9 de l'annexe II.
Conditions de l'essai.
Le demandeur doit solliciter au préalable l'agrément des autorités compétentes sur la nature et les conditions de l'essai à effectuer.
8.2.8. Plantes aquatiques.
Un essai sur les plantes aquatiques doit être effectué pour les herbicides.
Le demandeur doit solliciter au préalable l'agrément des autorités compétentes sur la nature et les conditions de l'essai à effectuer.
8.3. Effets sur les arthropodes.
8.3.1. Abeilles.
8.3.1.1. Toxicité aiguë.
Objet de l'essai.
L'essai devrait permettre d'établir les valeurs DL 50 concernant la toxicité aiguë orale ou de contact de la substance active.
Circonstances où l'essai est requis.
La détermination de l'impact potentiel sur les abeilles est obligatoire, sauf lorsque les préparations contenant la substance active sont exclusivement destinées à être utilisées dans des situations où l'exposition des abeilles est improbable, à savoir.
- l'entreposage des denrées alimentaires en espace clos ;
- le traitement non systémique des semences ;
- les préparations non systémiques pour l'épandage sur le sol ;
- les traitements non systémiques par trempage des plants et bulbes repiqués ;
- les traitements des plaies et des blessures ;
- les appâts rodenticides ;
- l'emploi en serre sans pollinisateur.
Lignes directrices.
L'essai doit être effectué en conformité avec la directive 170 de l'OEPP.
8.3.1.2. Essai d'alimentation du couvain d'abeilles.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer les risques éventuels que présente le produit phytopharmaceutique pour les larves d'abeilles.
Circonstances où l'essai est requis.
L'essai doit être réalisé lorsque la substance active peut agir comme régulateur de la croissance des insectes, sauf s'il est établi que l'exposition du couvain d'abeilles est improbable.
Lignes directrices.
L'essai doit être réalisé en conformité avec la méthode ICPBR (par exemple : P.A. Comen, A. de Ruijter et J. van der Steen. Method for honeybee brood feeding tests with insect growth-regulating insecticides. Bulletin OEPP, volume 22, 613-616, 1992).
8.3.2. Autres arthropodes.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer la toxicité (mortalité et effets sublétaux) de la substance active pour certaines espèces d'arthropodes.
Circonstances où l'essai est requis.
Il est obligatoire de déterminer les effets sur les arthropodes terrestres non cibles (par exemple, les prédateurs ou les parasitoïdes des organismes nuisibles). Les renseignements obtenus pour ces espèces peuvent également être utilisés pour indiquer la toxicité potentielle à l'égard d'autres espèces non cibles vivant dans le même environnement. Ces informations sont exigées pour toutes les substances actives, sauf lorsque les préparations contenant la substance active sont destinées exclusivement à être utilisées dans des situations où les arthropodes non cibles ne sont pas exposés, à savoir :
- l'entreposage de denrées alimentaires en espace clos ;
- les traitements des plaies et blessures ;
- les appâts rodenticides.
Conditions de l'essai.
L'essai doit d'abord être réalisé en laboratoire sur substrat artificiel (par exemple, plaque de verre ou sable de quartz, selon le cas), sauf si d'autres études révèlent la probabilité d'effets néfastes. Dans ce cas, des substrats plus adaptés peuvent être utilisés.
Deux espèces types sensibles, un parasitoïde et un acarien prédateur (par exemple, Aphidius rhopalosiphi et Typhlodromus pyri), sont à tester. A celles-là s'ajouteront deux espèces supplémentaires, qui doivent être appropriées à l'utilisation prévue de la substance.
Dans la mesure du possible et s'il y a lieu, elles représenteront les deux autres grands groupes en cause, les prédateurs vivant dans le sol et les prédateurs vivant dans le feuillage. Si des effets sont observés sur les espèces appropriées à l'utilisation envisagée pour le produit, d'autres essais peuvent être effectués en condition semi-naturelle. La sélection des espèces à soumettre à l'essai sera conforme aux propositions formulées dans le Guidance Document on Regulatory Testing Procedures for Pesticides with Non-Target ArthropodsWorkshop European Standard Characteristics of beneficials Regulatory Testing (ESCORT), 28-30 mars 1994. ISBN 0 9522535 2 6.
du Setac. Les essais utiliseront des doses équivalentes à la dose la plus élevée des applications recommandées sur le terrain.
Lignes directrices.
L'essai devrait être effectué en conformité avec les directives correspondantes qui répondent au moins aux exigences d'essai du Guidance Document on Regulatory Testing Procedures for Pesticides with Non-Target Arthropods du Setac.
8.4. Effets sur les vers de terre.
8.4.1. Toxicité aiguë.
Objet de l'essai.
L'essai devrait permettre d'établir la valeur de la CL 50 de la substance active à l'égard des vers de terre et, dans la mesure du possible, la concentration la plus élevée ne provoquant pas de mortalité et la concentration la plus faible provoquant 100 p. 100 de mortalité ; il mentionnera les effets observés sur la morphologie et le comportement.
Circonstances où l'essai est requis.
La détermination des effets sur les vers de terre est obligatoire lorsque les préparations contenant la substance active sont appliquées sur le sol ou sont susceptibles de contaminer le sol.
Lignes directrices.
L'essai doit être effectué en conformité avec la partie C :
Toxicité pour les vers de terre. Essai sur sol artificiel de la directive 88/302/CEE de la Commission, portant neuvième adaptation au progrès technique de la directive 67/548/CEE concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à la classification, l'emballage et l'étiquetage des substances dangereuses.
8.4.2. Effets sublétaux.
Objet de l'essai.
L'essai devrait permettre d'établir la CSEO et les effets sur la croissance, la reproduction et le comportement.
Circonstances où l'essai est requis.
Lorsque l'exposition continue ou répétée des vers de terre à la substance active ou à des quantités significatives de métabolites, de produits de dégradation ou de réaction est prévisible, sur la base du mode d'emploi proposé pour les préparations contenant la substance active ou sur la base du comportement et du sort de celle-ci dans le sol (TF 90 > 100 jours), un jugement d'expert est nécessaire pour décider si la détermination des effets sublétaux est utile.
Conditions de l'essai.
L'essai doit être réalisé sur Eiusenia foetida.
8.5. Effets sur les micro-organismes non cibles du sol.
Objet de l'essai.
L'essai devrait permettre d'obtenir des données suffisantes pour évaluer l'impact de la substance active sur l'activité microbienne du sol, traduite par la transformation de l'azote et la minéralisation du carbone.
Circonstances où l'essai est requis.
L'essai est obligatoire lorsque les préparations contenant la substance active sont appliquées sur le sol ou sont susceptibles de contaminer le sol dans les conditions d'utilisation courante. Dans le cas des substances actives destinées à être utilisées dans des préparations servant à la stérilisation des sols, les études doivent avoir pour objet la détermination des taux de récupération après traitement.
Conditions de l'essai.
Les sols utilisés doivent être des sols agricoles fraîchement échantillonnés. Les sites dont ils proviennent ne doivent pas avoir été traités au cours des deux années précédentes par des substances pouvant modifier considérablement la diversité et le niveau des populations microbiennes présentes à un titre autre que de manière transitoire.
Lignes directrices.
Setac. Procédures d'évaluation du comportement environnemental et de l'écotoxicité des pesticides.
8.6. Effets sur d'autres organismes non cibles (flore et faune) supposés être exposés à un risque.
Il est obligatoire de fournir un résumé des données résultant des essais préliminaires effectués pour évaluer l'activité biologique et les doses exploratrices, qu'elles soient positives ou négatives, de nature à fournir des renseignements sur l'impact éventuel d'autres espèces non cibles (flore et faune) et de les accompagner d'un avis critique sur la pertinence d'un impact potentiel sur les espèces non cibles.
8.7. Effets sur les méthodes biologiques de traitement des eaux usées.
Il est obligatoire de déterminer et consigner les effets sur les méthodes biologiques de traitement des eaux usées lorsque l'utilisation de produits phytopharmaceutiques contenant la substance active peut avoir des effets néfastes sur les installations de traitement des eaux usées.
9. Résumé et évaluation des points 7 et 8
(Arrêté du 1er février 1996, article 1er)
(Arrêté du 1er février 1996, article 1er)
Symbole(s) des dangers ;
Indications du danger ;
Phrases types relatives à la nature des risques ;
Phrases types relatives aux conseils de prudence.
11. Dossier conforme aux dispositions de l'annexe II, partie A, établi pour un produit phytopharmaceutique représentatif
(Arrêté du 1er février 1996, article 1er)
Partie B : Micro-organismes et virus
(Arrêté du 17 décembre 2002, article 1er)
Introduction
i) Les substances actives sont définies à l'article 2, paragraphe 4. Elles incluent les substances chimiques et les micro-organismes, y compris les virus. La présente partie énonce les informations requises pour les substances actives constituées de micro-organismes, y compris les virus. Aux fins de l'annexe I, partie B, le terme "micro-organisme" est utilisé dans l'acception suivante :
"Identité microbiologique, cellulaire ou non, capable de répliquer ou de transférer du matériel génétique. La définition s'applique, notamment mais pas exclusivement, aux bactéries, champignons, protozoaires, virus et viroïdes".
ii) Toute demande concernant des micro-organismes doit être accompagnée de toutes les informations et de toute la documentation pertinente disponible en l'état actuel des connaissances. Les informations les plus importantes et les plus utiles sont fournies par la caractérisation et l'identification du micro-organisme. Les informations de ce type sont définies dans les sections 1, 2 et 3 (identité, propriétés biologiques, informations complémentaires). Elles constituent la base de l'évaluation des effets du micro-organisme sur la santé humaine et sur l'environnement. Des données récentes issues d'expérimentations toxicologiques et/ou pathologiques sur des animaux de laboratoire sont normalement exigées, sauf si le demandeur est en mesure de démontrer, sur la base des informations précédemment fournies, que l'utilisation du micro-organisme, dans les conditions proposées, n'a aucun effet nocif sur la santé humaine ou animale, ni sur les eaux souterraines, et n'a aucune incidence inacceptable sur l'environnement.
iii) Dans l'attente de l'adoption de procédures spécifiques au niveau international, les informations requises seront obtenues en appliquant les procédures de test adoptées par l'autorité compétente (comme celles de l'USEPA, par exemple). Le cas échéant, il y a lieu d'adapter les procédures décrites à l'annexe I, partie A, pour qu'elles puissent convenir aux micro-organismes. Les tests doivent porter sur des micro-organismes viables, et, le cas échéant, non viables, et comporter un contrôle à blanc.
iv) Pour les tests effectués, il y a lieu de fournir une description détaillée (spécifications techniques) du matériel utilisé et des impuretés qu'il contient, conformément aux dispositions de la section 1, point 1.4. Le matériel utilisé doit correspondre aux spécifications techniques définies pour la fabrication des préparations à autoriser. Si des études sont effectuées avec des micro-organismes obtenus en laboratoire ou dans une installation pilote, elles doivent être répétées avec des micro-organismes obtenus industriellement, sauf s'il peut être démontré que le matériel utilisé est essentiellement le même aux fins des tests et des évaluations.
v) Dans le cas de micro-organismes génétiquement modifiés, il y a lieu de fournir une copie de l'évaluation des données relatives à l'estimation du risque pour l'environnement, comme prévu à l'article 30 du décret n° 94-359 du 5 mai 1994 susvisé.
vi) Le cas échéant, les données sont analysées à l'aide de méthodes statistiques appropriées. Les analyses statistiques doivent être rapportées de manière exhaustive (par exemple, toutes les estimations ponctuelles doivent être délimitées par un intervalle de confiance et il y a lieu de fournir les valeurs de probabilité exactes plutôt que d'utiliser la mention "significatif/non significatif".
vii) Dans le cas des études prévoyant une administration prolongée sur une certaine période, l'administration doit être effectuée de préférence au moyen d'un seul lot de micro-organismes, si la stabilité de celui-ci le permet. Si les études ne sont pas réalisées au moyen d'un lot unique de micro-organismes, il convient de certifier la similarité des différents lots utilisés. Si une étude comporte l'utilisation de doses différentes, la relation entre la dose et l'effet néfaste doit être notée.
viii) S'il est acquis que l'action de protection phytosanitaire est due à l'effet résiduel d'une toxine ou d'un métabolite, ou s'il faut s'attendre à la présence de résidus significatifs de toxines ou de métabolites non liés à l'effet de la substance active, ces toxines ou ces métabolites doivent faire l'objet d'un dossier constitué conformément aux prescriptions de l'annexe I, partie A.
1. Identité du micro-organisme
(Arrêté du 17 décembre 2002, article 1er)
L'identification ainsi que la caractérisation du micro-organisme fournissent les informations les plus importantes et constituent un élément clé de la prise de décision.
1.1. Demandeur.
Le nom et l'adresse du demandeur (adresse permanente dans la Communauté) doivent être indiqués, tout comme les nom, qualité, numéros de téléphone et de télécopieur de la personne à contacter. Lorsque, en outre, le demandeur dispose d'un bureau, d'un agent ou d'un représentant dans l'Etat membre auquel est présentée la demande d'inscription à l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques et, s'il est différent, dans l'Etat membre rapporteur nommé par la Commission, il convient d'indiquer le nom et l'adresse du bureau, de l'agent ou du représentant local, ainsi que les nom, position, numéros de téléphone et de télécopieur de la personne à contacter.
1.2. Producteur.
Le nom et l'adresse du ou des producteur(s) du micro-organisme doivent être fournis, de même que le nom et l'adresse de chaque unité assurant la production du micro-organisme. Indiquer un point de contact (de préférence central, avec nom, numéro de téléphone et numéro de télécopieur), auquel seront envoyées les données d'actualisation et où il sera répondu aux questions concernant la technologie de production, les procédés et la qualité du produit (y compris, le cas échéant, en ce qui concerne les lots individuels). Si la localisation ou le nombre des producteurs viennent à être modifiés après l'inscription de la substance active dans l'annexe de l'arrêté du 14 avril 1998 modifié établissant la liste des substances actives dont l'incorporation est autorisée dans les produits phytopharmaceutiques, l'information requise doit être à nouveau notifiée à la Commission et aux Etats membres.
1.3. Nom et description des espèces, caractérisation de la souche.
i) Le micro-organisme doit être déposé auprès d'une banque de collection de cultures de réputation internationale. Indiquer le numéro de dépôt correspondant, ainsi que les coordonnées de l'institution.
ii) Chacun des micro-organismes visés par la demande doit être identifié et désigné par son nom d'espèce. Il convient d'indiquer le nom scientifique et le groupe taxonomique, c'est-à-dire la famille, le genre, l'espèce, la souche, le sérotype, le pathovar et toute autre dénomination pertinente de chaque micro-organisme. Préciser si le micro-organisme :
- est indigène ou non indigène, au niveau de l'espèce, du domaine d'application proposé ;
- est une souche sauvage ;
- est un mutant spontané ou induit ;
- a été modifié au moyen de techniques décrites aux annexes du décret n° 93-774 du 27 mars 1993 susvisé.
Dans ces deux derniers cas, il convient d'indiquer toutes les différences connues entre le micro-organisme modifié et la souche sauvage initiale.
iii) La technologie la plus avancée disponible doit être utilisée pour identifier et caractériser le micro-organisme au niveau de la souche. Indiquer les procédures de test et les critères utilisés pour l'identification (par exemple, morphologie, biochimie, sérologie, identification moléculaire).
iv) Indiquer le nom commun ainsi que, le cas échéant, tout nom supplémentaire, de remplacement ou de code utilisés lors de la phase de développement.
v) Indiquer toute parenté avec des pathogènes connus.
1.4. Spécifications techniques du matériel utilisé pour la fabrication des produits formulés.
1.4.1. Teneur en micro-organisme.
La teneur minimale et maximale en micro-organisme du matériel utilisé pour la fabrication des produits formulés doit être indiquée, et ce en termes appropriés, tels que le nombre d'unités actives par unité de volume ou de poids, ou de toute autre manière adéquate pour le micro-organisme considéré. Lorsque l'information fournie concerne un système de production pilote, l'information requise doit de nouveau être fournie à la Commission et aux Etats membres lorsque les méthodes et les procédures de production à l'échelle industrielle se sont stabilisées et si les changements intervenus dans la production modifient les spécifications en termes de pureté.
1.4.2. Identité et teneur des impuretés et des additifs contaminant les micro-organismes.
Dans toute la mesure possible, il est souhaitable que les produits phytopharmaceutiques soient exempts de contaminants (y compris les micro-organismes contaminants). Le niveau et la nature des contaminants acceptables doivent être établis par l'autorité compétente sur la base d'une évaluation des risques. Dans la mesure du possible, il convient d'indiquer, le cas échéant, l'identité et la teneur maximale, exprimée dans l'unité appropriée, de tous les micro-organismes contaminants. Chaque fois que possible, les données relatives à l'identité doivent être fournies conformément aux exigences de l'annexe I, partie B, section 1, point 1.3. Il convient d'identifier et de caractériser, à différents états ou stades de croissance du micro-organisme, tout métabolite sensible (c'est-à-dire susceptible de poser un risque pour la santé humaine ou pour l'environnement) produit par le micro-organisme (introduction, point viii), de l'annexe I B). Fournir, s'il y a lieu, des informations détaillées sur tous les composants tels que les condensés, milieux de culture, etc.
En ce qui concerne les impuretés chimiques sensibles pour la santé humaine ou pour l'environnement, indiquer leur identité et leur teneur maximale, exprimée dans l'unité appropriée. En ce qui concerne les additifs, indiquer leur identité et leur teneur, exprimée en grammes par kilogramme. Les données relatives à l'identité des substances chimiques telles que les additifs doivent être fournies comme prescrit à l'annexe I, partie A, section 1, point 1.10.
1.4.3. Profil analytique des lots.
Fournir, s'il y a lieu, les données visées à l'annexe I, partie A, section 1, point 1.11, dans les unités appropriées.
2. Propriétés biologiques du micro-organisme
(Arrêté du 17 décembre 2002, article 1er)
2.1. Historique du micro-organisme et de ses utilisations. Présence naturelle et répartition géographique.
Il convient de présenter la familiarité du micro-organisme, c'est-à-dire la disponibilité de connaissances appropriées le concernant.
2.1.1. Historique.
Présenter l'historique du micro-organisme et de ses utilisations (expériences et projets de recherche ou utilisations commerciales).
2.1.2. Origine et présence naturelle.
Indiquer la région géographique et la situation dans l'écosystème (par exemple, la plante hôte, l'animal hôte, ou encore le sol dans lequel le micro-organisme a été isolé), en précisant le mode d'isolement utilisé. La présence naturelle du micro-organisme dans l'environnement concerné doit être indiquée, si possible, au niveau de la souche.
Dans le cas d'un micro-organisme mutant ou génétiquement modifié (au sens du décret n° 93-774 susvisé), fournir une description détaillée du mode de production et d'isolement, ainsi que des moyens permettant de le distinguer clairement de la souche sauvage initiale.
2.2. Informations sur l'organisme cible/les organismes cibles.
2.2.1. Description de l'organisme cible/des organismes cibles.
Le cas échéant, spécifier les organismes nuisibles contre lesquels une protection est assurée.
2.2.2. Mode d'action.
Indiquer le mode d'action principal. En liaison avec le mode d'action, préciser également si le micro-organisme produit une toxine ayant un effet résiduel sur l'organisme cible. Si c'est le cas, décrire le mode d'action de cette toxine. Le cas échéant, fournir des informations sur le site d'infection, le mode d'entrée dans l'organisme cible et ses phases sensibles. Les résultats de toute étude expérimentale doivent être communiqués. II convient de préciser les voies possibles d'inoculation du micro-organisme ou de ses métabolites, et spécialement des toxines (contact, ingestion, inhalation). Préciser également si le micro-organisme ou ses métabolites sont transportés dans des végétaux et, si c'est le cas, comment ce déplacement a lieu. En cas d'effet pathogène sur l'organisme cible, préciser la dose infectante (dose nécessaire pour infecter une espèce cible avec l'effet souhaité) et la transmissibilité du micro-organisme (faculté de diffusion dans la population cible, mais également d'une espèce cible à une autre espèce "cible") après application dans les conditions d'utilisation proposées.
2.3. Gamme de spécificité de l'hôte et effets sur des espèces autres que l'organisme nuisible cible.
Fournir toute information disponible en ce qui concerne les effets sur les organismes autres que l'organisme cible dans le secteur où le micro-organisme peut se propager, en signalant également la présence de tout organisme autre que l'organisme cible qui serait soit très proche de l'espèce cible, soit particulièrement exposé. Toute connaissance concernant la toxicité de la substance active ou de ses métabolites pour les humains ou les animaux, sa capacité éventuelle à coloniser ou à infester des humains ou des animaux (y compris les sujets immunodéprimés) et ses éventuels effets pathogènes doit être mentionnée. Il convient également de signaler tout élément connu permettant d'indiquer si la substance active ou ses dérivés sont irritants pour la peau, les yeux ou les organes respiratoires des personnes humaines ou des animaux et s'ils peuvent entraîner des réactions allergiques en cas de contact avec la peau ou d'inhalation.
2.4. Phase de développement/cycle de vie du micro-organisme.
Il convient de présenter toute information disponible sur le cycle de vie du micro-organisme, les cas décrits de symbiose, de parasitisme et de concurrence, les prédateurs, etc., ainsi que les organismes hôtes. Pour les virus, signaler également les vecteurs. Indiquer le temps de génération et le type de reproduction du micro-organisme, de même que les données relatives aux éventuelles phases de repos du micro-organisme, à sa durée de vie, à sa virulence et à son potentiel d'infection. Spécifier également si le micro-organisme, au cours des différentes phases de développement suivant sa libération, possède la faculté de produire des métabolites, notamment des toxines, susceptibles de présenter un risque pour la santé humaine ou pour l'environnement.
2.5. Infectiosité, capacité de propagation, capacité de colonisation.
Indiquer la persistance du micro-organisme et fournir les renseignements relatifs à son cycle de vie dans les conditions environnementales caractéristiques de l'utilisation prévue. Signaler en outre toute sensibilité particulière du micro-organisme à certaines composantes environnementales (rayons ultraviolets, sols, eau). Indiquer les conditions environnementales (température, pH, humidité, nutriments, etc.) nécessaires à la survie et à la reproduction du micro-organisme, ainsi qu'à sa capacité de colonisation et de destruction (notamment des tissus humains) et à son efficacité. La présence de facteurs spécifiques de virulence doit être mentionnée.
Il convient de déterminer la fourchette de températures dans laquelle la croissance du micro-organisme est possible, en précisant les températures minimale, maximale et optimale. Ces données sont particulièrement utiles pour conduire l'étude des effets sur la santé humaine (section 5). Indiquer également l'influence éventuelle de facteurs tels que la température, les rayons ultraviolets, le pH et la présence de certaines substances sur la stabilité des toxines concernées. Fournir toute information relative aux itinéraires possibles de propagation du micro-organisme (par le biais de poussières en suspension dans l'air, d'organismes hôtes jouant le rôle de vecteurs, etc.), dans les conditions environnementales caractéristiques de l'utilisation prévue.
2.6. Parenté avec des pathogènes connus des plantes, des animaux ou des personnes humaines.
Indiquer l'existence éventuelle d'une ou de plusieurs espèces du même genre que les micro-organismes actifs et/ou, le cas échéant, contaminants qui ont un effet pathogène connu sur les êtres humains, les animaux, les cultures ou des espèces non visées. Préciser les types de pathologies causées. Spécifier s'il est possible de distinguer sans ambiguïté le micro-organisme actif des espèces pathogènes (et dans ce cas, par quel moyen).
2.7. Stabilité génétique du micro-organisme et facteurs susceptibles de l'affecter.
Fournir, s'il y a lieu, des informations sur la stabilité génétique du micro-organisme (taux de mutation des traits relatifs au mode d'action, par exemple, ou absorption de matériel génétique exogène), dans les conditions environnementales de l'utilisation proposée.
Fournir également des informations sur la capacité du micro-organisme à transférer du matériel génétique vers d'autres organismes et son potentiel pathogène pour les végétaux, les animaux et les êtres humains. Si le micro-organisme est porteur d'éléments génétiques sensibles supplémentaires, préciser la stabilité des traits encodés.
2.8. Informations relatives à la production de métabolites (et particulièrement de toxines).
Lorsqu'il est connu que d'autres souches de la même espèce microbienne que la souche objet de la demande a la faculté de produire des métabolites (et spécialement des toxines) dont les effets sur la santé humaine et/ou sur l'environnement, en cours d'application ou après l'application, sont notoirement inacceptables, il y a lieu de décrire la nature et la structure de la substance en cause, son mode d'action (en précisant les facteurs internes et externes nécessaires à l'action du micro-organisme), ainsi que ses effets sur les êtres humains, les animaux ou d'autres espèces non visées. Il convient en outre de décrire les conditions de production des métabolites (et notamment des toxines) par le micro-organisme, en incluant toutes les informations disponibles sur le mécanisme de régulation de la production des métabolites ainsi que l'influence des métabolites produits sur le mode d'action du micro-organisme.
2.9. Antibiotiques et autres agents antimicrobiens.
De nombreux micro-organismes produisent certaines substances antibiotiques. A toutes les étapes de l'élaboration d'un produit phytopharmaceutique microbien, il est impératif d'éviter les interférences avec l'utilisation des antibiotiques en médecine humaine ou vétérinaire. Il convient en conséquence de fournir des informations sur la résistance ou la sensibilité du micro-organisme aux antibiotiques comme à d'autres agents antimicrobiens, notamment en ce qui concerne la stabilité des codes génétiques déterminant la résistance aux antibiotiques, sauf s'il peut être démontré que le micro-organisme n'a aucun effet nuisible sur la santé humaine ou animale, ou ne possède pas la faculté de transférer sa résistance aux antibiotiques et autres agents antimicrobiens.
(Arrêté du 17 décembre 2002, article 1er)
Introduction.
i) L'information fournie doit indiquer à quelles fins on utilise ou prévoit d'utiliser les préparations contenant le micro-organisme et préciser les doses et les modes d'utilisation pratiqués ou proposés.
ii) L'information fournie doit préciser les méthodes et les précautions ordinaires à observer dans la manipulation, l'entreposage et le transport du micro-organisme.
iii) Les études, les données et les informations présentées doivent démontrer que les mesures proposées conviennent dans des situations d'urgence.
iv) Les informations et les données visées sont requises pour tous les micro-organismes, sauf indication contraire.
3.1. Fonction.
Préciser la fonction biologique à retenir parmi les suivantes :
- bactéricide ;
- fongicide ;
- insecticide ;
- acaricide ;
- molluscicide ;
- nématicide ;
- herbicide ;
- autres (à préciser).
3.2. Domaine d'utilisation envisagé.
Indiquer, dans la liste ci-après, le ou les domaines d'utilisation actuels et proposés des préparations contenant le micro-organisme :
- utilisation sur le terrain (agriculture, horticulture, forêt, viticulture, etc.) ;
- cultures protégées (sous serre, par exemple) ;
- cultures ornementales ;
- désherbage des terres non cultivées ;
- jardinage ;
- plantes d'intérieur ;
- produits stockés ;
- autres (préciser).
3.3. Cultures ou produits protégés ou traités.
Préciser l'utilisation actuelle et envisagée en termes de cultures, groupes de cultures, végétaux ou produits végétaux protégés.
3.4. Mode de production et contrôle de qualité.
Fournir une description exhaustive du mode de production à grande échelle du micro-organisme. Le demandeur doit assurer un contrôle de qualité continu tant du processus ou de la méthode de production que du produit obtenu. Il importe notamment de surveiller toute modification spontanée des principales caractéristiques du micro-organisme ainsi que la présence ou l'absence de contaminants significatifs.
Soumettre le détail des critères de garantie de la qualité applicables à la production.
Décrire et spécifier les techniques employées pour garantir l'uniformité du produit et les méthodes de titrage utilisées pour assurer la standardisation, la conservation et la pureté du micro-organisme (par exemple, HACCP).
3.5. Informations relatives à l'apparition ou à l'apparition éventuelle du développement d'une résistance de l'organisme cible/des organismes cibles.
Fournir toute information disponible sur l'apparition éventuelle d'une résistance ou d'une résistance croisée de l'organisme cible/des organismes cibles.
Décrire, le cas échéant, les stratégies de réponse appropriées.
3.6. Méthodes employées pour empêcher la perte de virulence du stock de semence du micro-organisme.
Décrire les méthodes destinées à empêcher la perte de virulence des cultures initiales. Décrire en outre toute méthode éventuellement disponible pour éviter que le micro-organisme ne perde son efficacité sur les espèces cibles.
3.7. Procédures et précautions recommandées en matière de manipulation, d'entreposage et de transport ou en cas d'incendie.
Pour chaque micro-organisme, fournir une fiche de données de sécurité semblable à celle requise pour les substances au titre des articles R. 231-53 et R. 231-53-1 du code du travail.
3.8. Procédures de destruction ou de décontamination.
Dans de nombreux cas, le meilleur ou l'unique moyen d'éliminer en toute sécurité des micro-organismes, des matières contaminées ou des emballages contaminés est de les soumettre à une incinération contrôlée dans un incinérateur agréé. Fournir une description exhaustive des méthodes employées pour éliminer en toute sécurité le micro-organisme, ou, s'il y a lieu, pour le tuer avant élimination, ainsi que les modes d'élimination des emballages et des matières contaminées. Fournir des données permettant d'établir l'efficacité et la sûreté de ces méthodes.
3.9. Mesures en cas d'accident.
Décrire les procédures destinées à rendre le micro-organisme inoffensif dans l'environnement (par exemple l'eau ou le sol) en cas d'accident.
Article Annexe I
Créé par JORF 14 janvier 2003
4. Méthodes d'analyse
(Arrêté du 17 décembre 2002, article 1er)
Introduction.
Les dispositions de la présente section s'appliquent exclusivement aux méthodes d'analyse requises pour le contrôle et le suivi postérieurs à l'autorisation.
Pour tous les éléments d'évaluation des risques, un suivi post-autorisation pourra être envisagé, notamment lorsqu'une demande concerne des (souches) de micro-organismes qui ne sont pas indigènes de la zone d'application envisagée. En ce qui concerne les méthodes d'analyse utilisées pour la production des données requises par le présent arrêté ou à d'autres fins, le demandeur est tenu de fournir une justification de la méthode utilisée. Si nécessaire, des arrêtés spécifiques seront élaborés pour ces méthodes sur la base des mêmes normes que celles requises pour les méthodes de contrôle et de suivi postérieurs à l'autorisation. Il convient de fournir une description des méthodes d'analyse contenant toutes les données utiles relatives à l'équipement et au matériel utilisés ainsi qu'aux conditions d'application. L'application de toute méthode internationalement reconnue doit être signalée.
Ces méthodes doivent, autant que possible, suivre l'approche la plus simple, être peu onéreuses et faire appel à des équipements courants. Pour les méthodes d'analyse des micro-organismes et de leurs résidus, il y a également lieu de fournir les données relatives à la spécificité, à la linéarité, à l'exactitude et à la répétabilité, telles que définies à l'annexe I, partie A, points 4.1 et 4.2.
Les définitions visées ci-après s'appliquent aux fins de la présente section.
Impuretés : tous les composants (y compris les micro-organismes contaminants et/ou les substances chimiques) autres que le micro-organisme désigné, provenant du processus de fabrication ou d'une dégradation intervenue en cours de stockage.
Impuretés sensibles : impuretés, telles que définies ci-dessus, qui présentent un risque pour la santé humaine ou animale et/ou pour l'environnement.
Métabolites : produits résultant notamment de réactions biosynthétiques et de dégradation intervenant au sein du micro-organisme ou d'autres organismes utilisés pour la production du micro-organisme concerné.
Métabolites sensibles : métabolites qui présentent un risque pour la santé humaine ou animale et/ou pour l'environnement
Résidus : micro-organismes viables et substances fabriquées en quantité significative par les micro-organismes, qui persistent après la disparition des micro-organismes et présentent un risque pour la santé humaine ou animale et/ou l'environnement.
Les échantillons suivants doivent être fournis sur demande :
i) Des échantillons du micro-organisme tel qu'il est produit ;
ii) Des étalons pour l'analyse des métabolites sensibles (spécialement des toxines) et de tous les autres composants compris dans la définition des résidus ;
iii) Si disponibles, des échantillons des substances de référence des impuretés sensibles.
4.1. Méthodes d'analyse du micro-organisme tel qu'il est produit Méthodes d'identification du micro-organisme.
Méthodes d'obtention d'informations sur la variabilité possible du stock de semence/du micro-organisme actif.
Méthodes employées pour différencier les mutants du micro-organisme de la souche sauvage initiale.
Méthodes employées pour établir la pureté du stock de semences à partir duquel les lots sont produits et méthodes employées pour contrôler la pureté.
Méthodes employées pour déterminer la teneur en micro-organisme du matériel manufacturé utilisé pour la production des produits formulés et méthodes permettant de démontrer que les micro-organismes contaminants sont contenus dans des limites acceptables.
Méthodes d'identification des impuretés sensibles dans le matériel manufacturé.
Méthodes employées pour vérifier l'absence ou quantifier la présence (avec les limites appropriées de détermination) de tout agent pathogène pour les êtres humains et les mammifères.
Méthodes permettant de déterminer la stabilité de stockage et la durée de conservation du micro-organisme, le cas échéant.
4.2. Méthodes de détermination et de quantification des résidus (viables ou non viables).
Du/des micro-organisme(s) actif(s).
Des métabolites sensibles (spécialement les toxines), présents sur et/ou dans les cultures, dans les denrées alimentaires et les aliments pour animaux, dans les tissus et les fluides des êtres humains et des animaux, dans les sols, dans l'eau (à savoir l'eau potable, l'eau souterraine et l'eau de surface) ainsi que dans l'air, selon le cas.
Il convient d'inclure les méthodes analytiques de détermination de la teneur ou de l'activité des produits protéiques, telles que l'analyse des cultures exponentielles et des surnageants dans un essai biologique de culture de cellules animales.
5. Effets sur la santé des personnes
(Arrêté du 17 décembre 2002, article 1er)
Introduction.
i) Les informations disponibles sur les propriétés du micro-organisme et des organismes concernés (sections 1 à 3), y compris les rapports sanitaires et médicaux, peuvent suffire pour déterminer si le micro-organisme est ou non susceptible d'avoir un effet (infectieux, pathogène ou toxique) sur la santé humaine.
ii) Les informations fournies, jointes à celles concernant une ou plusieurs préparations contenant le micro-organisme, doivent être suffisantes pour permettre une évaluation des risques pour les personnes découlant, directement ou indirectement, de la manipulation et de l'utilisation de produits phytopharmaceutiques contenant le micro-organisme, ainsi que du risque pour les personnes lié aux résidus ou aux contaminants contenus dans les aliments et dans l'eau. En outre, les informations fournies doivent permettre :
- d'arrêter une décision quant à l'inclusion éventuelle de la substance active dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- de spécifier les conditions ou les restrictions appropriées devant accompagner toute inclusion dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- de décider des avertissements liés aux risques et aux normes de sécurité (une fois introduits) concernant la protection des personnes, des animaux et de l'environnement à inscrire sur les emballages (récipients) ;
- de définir les soins d'urgence appropriés ainsi que les mesures adéquates de diagnostic et de traitement thérapeutique à appliquer aux personnes en cas d'infection ou d'autre effet nocif.
iii) Tous les effets constatés au cours des recherches doivent être mentionnés. Il convient également d'engager les recherches qui peuvent être nécessaires afin d'identifier le mécanisme probablement à l'origine des effets constatés et d'évaluer la gravité de ces effets.
iv) Pour toutes les études, la dose réelle employée, exprimée en unités formant colonies par kilogramme de poids corporel, ainsi que dans d'autres unités appropriées, doit être mentionnée.
v) L'évaluation du micro-organisme doit être effectuée par phases.
La première phase concerne les informations de base disponibles et les études de base, qui doivent être réalisées pour tous les micro-organismes. Il revient aux experts de décider cas par cas du programme de tests approprié. Des données récentes issues d'expérimentations toxicologiques et/ou pathologiques sur des animaux de laboratoire sont normalement exigées, sauf si le demandeur est en mesure de démontrer, sur la base des informations précédemment fournies, que l'utilisation du micro-organisme, dans les conditions proposées, n'a aucun effet nocif sur la santé humaine ou animale. Dans l'attente de l'adoption de procédures spécifiques au niveau international, les informations requises sont obtenues en appliquant les procédures de test disponibles (USEPA OPPTS, par exemple).
Une deuxième phase d'études doit être menée si les tests de la première phase mettent au jour des effets nocifs sur la santé. Le type d'études à réaliser dépend de la nature des effets en question. Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
Phase I.
5.1. Informations de base.
Des informations de base doivent être fournies sur les éventuels effets nocifs du micro-organisme, à savoir notamment sa capacité à former des colonies, à causer des dommages et à produire des toxines et autres métabolites sensibles.
5.1.1. Données médicales :
Si elles sont disponibles, et sans préjudice des dispositions des articles R. 231-62 à R. 231-65-3 du code du travail concernant la protection des travailleurs contre les risques liés à l'exposition à des agents biologiques au travail, les données et les informations pratiques concernant la reconnaissance des symptômes d'infection ou de pathogénicité et l'efficacité des premiers soins et des mesures thérapeutiques doivent être présentées.
S'il y a lieu, l'efficacité d'antidotes potentiels doit être étudiée et relatée et les méthodes permettant de tuer ou d'inactiver le micro-organisme doivent être indiquées (section 3, point 3.8).
Les données et les informations concernant les effets de l'exposition humaine, pour autant qu'elles soient disponibles au niveau de qualité nécessaire, ont une valeur particulière parce qu'elles peuvent confirmer le bien-fondé des extrapolations et des conclusions relatives aux organes cibles, à la virulence et à la réversibilité des effets nocifs. De telles données peuvent être recueillies à la suite d'expositions résultant d'accidents ou d'activités professionnelles.
5.1.2. Surveillance médicale du personnel de l'établissement.
Les rapports disponibles des programmes de surveillance de la médecine du travail, étayés d'informations détaillées sur la conception du programme et l'exposition au micro-organisme, doivent être soumis. Ces rapports doivent comprendre, dans la mesure du possible, des informations relatives au mécanisme d'action du micro-organisme. De même, ils doivent comporter les données éventuellement disponibles concernant les personnes exposées dans les usines de production ou après application du micro-organisme (par exemple, dans le cadre de tests d'efficacité).
Il convient d'accorder une attention particulière aux personnes dont la sensibilité peut être affectée, par exemple, par une maladie préexistante, un médicament, un système immunitaire fragilisé, la grossesse ou l'allaitement.
5.1.3. Observations éventuelles de sensibilisation/pouvoir allergisant.
Il convient de fournir toute information disponible sur des cas de sensibilisation et de réaction allergique chez les professionnels, à savoir les travailleurs des usines, les travailleurs agricoles, les chercheurs et toute autre personne exposée au micro-organisme, en joignant, le cas échéant, une description détaillée de toute incidence d'hypersensibilité et de sensibilisation chronique. Les informations fournies doivent comporter des détails sur la fréquence, le niveau et la durée de l'exposition, les symptômes observés et les autres observations cliniques pertinentes. Il convient également de préciser si les professionnels concernés ont subi des tests allergiques ou ont été interrogés sur des manifestations allergiques.
5.1.4. Observation directe (cas cliniques, par exemple).
Il convient de fournir les rapports provenant de sources bibliographiques publiques relatifs aux cas cliniques concernant le micro-organisme ou des membres étroitement apparentés du même groupe taxonomique, s'ils sont issus de revues autorisées ou de rapports officiels, ainsi que tout rapport concernant d'éventuelles études de suivi. Ces rapports, particulièrement utiles, doivent comporter des descriptions exhaustives de la nature, du degré et de la durée de l'exposition ainsi que la mention des symptômes cliniques observés, des premiers soins et des actions thérapeutiques appliqués, des données mesurées et des observations effectuées. Un résumé ou des informations succinctes présentent peu d'intérêt.
Dans le cas où des études ont été réalisées sur l'animal, les rapports relatifs aux cas cliniques peuvent être particulièrement utiles pour confirmer la validité des extrapolations de l'animal à l'être humain et identifier tout effet nocif inattendu spécifique aux personnes humaines.
5.2. Etudes de base.
Pour pouvoir interpréter correctement les résultats obtenus, il est de la plus haute importance que les méthodes de test proposées soient appropriées en ce qui concerne la sensibilité, le mode d'administration, etc., et soient également adaptées du point de vue biologique et toxicologique. Le mode d'administration du micro-organisme utilisé aux fins de test est fonction des principaux types d'exposition des personnes.
Afin d'évaluer les effets à moyen et à long terme d'une exposition aiguë, subaiguë ou semi-chronique au micro-organisme, il est obligatoire d'appliquer la procédure figurant dans la plupart des orientations de l'OCDE, qui consiste à compléter les études réalisées par une période de récupération à l'issue de laquelle on effectue une analyse pathologique macro-et microscopique complète, avec recherche exploratoire du micro-organisme dans les tissus et les organes. Il est ainsi possible de faciliter l'interprétation de certains faits et d'établir l'infectiosité ou la pathogénicité, ce qui permet en retour de prendre des décisions sur d'autres points, tels que la nécessité d'entreprendre des études à long terme (de cancérogenèse, etc., comme évoqué au point 5.3) ou l'opportunité d'effectuer ou non des études sur les résidus (voir point 6.2).
5.2.1. Sensibilisation.
Les méthodes disponibles pour tester la sensibilisation cutanée ne sont pas appropriées dans le cas des micro-organismes. La sensibilisation par inhalation pose très probablement de plus grands problèmes que l'exposition cutanée aux micro-organismes, mais aucune méthode de test n'a jusqu'ici été validée. Le développement de ces types de méthodes revêt donc une grande importance. Jusque-là, il conviendra de considérer tous les micro-organismes comme des sensibilisateurs potentiels. Cette approche tient aussi compte des personnes immunodéprimées ou fragiles (telles que les femmes enceintes, les nourrissons ou les personnes âgées).
Objet du test :
Le test vise à fournir des informations suffisantes pour évaluer la capacité du micro-organisme à induire des réactions de sensibilisation par inhalation et par exposition cutanée. Il y a lieu d'effectuer un test maximisé.
Situations dans lesquelles les tests sont requis faute de méthodes de test appropriées, tous les micro-organismes sont considérés comme des sensibilisateurs potentiels, à moins que le demandeur ne s'efforce de prouver l'absence de potentiel de sensibilisation en présentant les données correspondantes. La soumission de telles données présente donc provisoirement un caractère non pas obligatoire mais facultatif.
Toute information relative à la sensibilisation doit être rapportée.
5.2.2. Toxicité, pathogénicité et infectiosité aiguës.
Les études, données et informations à fournir et à évaluer doivent être suffisantes pour permettre de déceler les effets d'une exposition unique au micro-organisme, et en particulier d'établir ou d'indiquer :
- la toxicité, la pathogénicité et l'infectiosité du micro-organisme ;
- l'évolution au cours du temps et les caractéristiques des effets, avec description exhaustive des modifications comportementales et des éventuelles constatations macropathologiques à l'inspection post mortem ;
- si possible, le mode d'action toxique ;
- les risques relatifs liés aux diverses voies d'exposition ;
- les analyses de sang réalisées au cours de toutes les études, afin d'évaluer l'élimination du micro-organisme.
Les effets toxiques/pathogènes aigus peuvent être accompagnés d'une infectiosité et/ou d'autres effets à long terme qui ne peuvent être observés immédiatement. En vue de l'évaluation sanitaire, il est donc nécessaire d'étudier la capacité d'infection par ingestion, inhalation et injection intrapéritonéale/sous-cutanée sur des mammifères de laboratoire.
Les études de toxicité, de pathogénicité et d'infectiosité aiguës doivent comporter une évaluation de l'élimination du micro-organisme et/ou de la toxine active dans les organes jugés appropriés pour l'examen microbien (par exemple, le foie, les reins, la rate, les poumons, le cerveau, le sang et le site d'administration).
Les observations à faire doivent refléter un jugement scientifique d'expert et peuvent inclure un décompte du micro-organisme dans tous les tissus susceptibles d'être touchés (présentant des lésions, par exemple) et dans les organes vitaux : reins, cerveau, foie, poumons, rate, vessie, sang, ganglions lymphatiques, appareil gastro-intestinal, thymus ainsi qu'au niveau des lésions au site d'inoculation chez les animaux morts ou moribonds, en cours de test et au moment du sacrifice de l'animal.
Les informations produites par les tests de toxicité, de pathogénicité et d'infectiosité aiguës sont particulièrement utiles pour évaluer les risques susceptibles de se présenter en cas d'accident ainsi que les risques pour le consommateur en cas d'exposition à d'éventuels résidus.
5.2.2.1. Toxicité par voie orale, pathogénicité et infectiosité aiguës.
Situations dans lesquelles les tests sont requis :
La toxicité aiguë par voie orale ainsi que la pathogénicité et l'infectiosité aiguës du micro-organisme doivent être signalées.
5.2.2.2. Toxicité aiguë par inhalation ; pathogénicité et infectiosité aiguës.
Situations dans lesquelles les tests sont requis :
La toxicité aiguë par inhalation. L'étude d'inhalation peut être remplacée par une étude intratrachéale. ainsi que la pathogénicité et l'infectiosité aiguës du micro-organisme doivent être signalées.
5.2.2.3. Dose unique intrapéritonéale/sous-cutanée.
Le test intrapéritonéal/sous-cutané est considéré comme un mode hautement sensible de mise en évidence, notamment, de l'infectiosité.
Situation dans lesquelles les tests sont requis :
L'injection intrapéritonéale est systématiquement requise pour tous les micro-organismes. Toutefois, dans le cas où leur température maximale de croissance et de multiplication serait inférieure à 37° C, il est laissé à l'appréciation des experts de décider s'il est préférable de substituer une injection sous-cutanée à l'injection intrapéritonéale.
5.2.3. Tests de génotoxicité.
Situation dans lesquelles les tests sont requis :
Si le micro-organisme produit des exotoxines au sens du point 2.8, ces toxines et tout autre métabolite sensible présent dans le milieu de culture doivent aussi être soumis à des tests de génotoxicité pratiqués, si possible, sur une forme purifiée de la substance chimique.
Lorsque les études de base ne révèlent pas la formation de métabolites toxiques, il convient d'examiner le micro-organisme lui-même en se fondant sur les avis des experts concernant l'importance et la validité des données de base. Dans le cas des virus, il y a lieu d'examiner le risque de mutagenèse insertionnelle dans les cellules de mammifères et le risque de cancérogenèse.
Objet du test :
Ces études présentent un intérêt pour :
- la prédiction du pouvoir génotoxique ;
- l'identification précoce des cancérogènes génotoxiques ;
- l'explication du mécanisme d'action de certains cancérogènes.
Il importe d'adopter une attitude souple, les autres tests à réaliser devant être fonction de l'interprétation des résultats à chaque étape.
Modalités des tests. Les méthodes de test actuelles étant conçues pour les substances chimiques solubles, il est nécessaire de les adapter aux micro-organismes.
La génotoxicité des micro-organismes cellulaires doit être étudiée, dans la mesure du possible, après la partition des cellules. Il convient de décrire la méthode de préparation de l'échantillon. La génotoxicité des virus doit être étudiée sur des isolats infectieux.
5.2.3.1. Tests in vitro.
Situations dans lesquelles les tests sont requis :
Il convient de fournir les résultats des tests de mutagenèse in vitro (essai bactérien relatif à la mutation génique, test de clastogénicité dans des cellules de mammifères et test de mutation génique dans des cellules de mammifères).
5.2.4. Etude de cultures de cellules.
Cette information est requise pour les micro-organismes intracellulaires réplicables tels que les virus, les viroïdes, certaines bactéries et certains protozoaires, sauf dans les cas où il ressort clairement des informations prévues aux sections 1 et 3 que les micro-organismes concernés ne se répliquent pas dans les organismes à sang chaud. L'étude à réaliser doit porter sur des cultures de cellules ou de tissus humains provenant de différents organes, sélectionnés par exemple sur la base des organes potentiellement visés par l'infection. Si les cultures de cellules ou de tissus humains provenant d'organes spécifiques ne sont pas disponibles, il est possible d'utiliser des cultures de cellules et de tissus provenant d'autres mammifères. En ce qui concerne les virus, la capacité d'interagir avec le génome humain est un élément clé.
5.2.5. Informations sur la toxicité et la pathogénicité à court terme.
Objet du test :
Les études de toxicité à court terme doivent être conçues pour fournir des informations sur la quantité de micro-organisme pouvant être tolérée sans provoquer d'effets toxiques dans les conditions de l'étude. Ces études fournissent des données utiles sur les risques encourus par les personnes qui manipulent et utilisent des préparations contenant le micro-organisme. En particulier, les études à court terme donnent un aperçu déterminant des effets cumulés possibles du micro-organisme et des risques encourus par les travailleurs qui y sont exposés de façon intensive. En outre, elles fournissent des informations utiles pour la conception des études de toxicité chronique.
Les études, données et informations à fournir et à évaluer doivent être suffisantes pour permettre de déceler les effets découlant d'une exposition répétée au micro-organisme et, en outre, d'établir ou d'indiquer notamment :
- la relation entre la dose et les effets néfastes ;
- la toxicité du micro-organisme, y compris le cas échéant le NOAEL (niveau sans effet négatif visible) correspondant aux toxines ;
- les organes cibles, le cas échéant ;
- l'évolution au cours du temps et les caractéristiques des effets, avec description exhaustive des modifications comportementales et des éventuelles constatations pathologiques à l'inspection post mortem ;
- les effets toxiques particuliers et les changements pathologiques provoqués ;
- le cas échéant, la persistance et la réversibilité de certains effets toxiques observés à la suite d'une interruption d'administration ;
- si possible, le mode d'action toxique ; ainsi que
- les risques relatifs liés aux diverses voies d'exposition.
Une estimation de l'élimination du micro-organisme dans les organes principaux doit être effectuée au cours de l'étude de toxicité à court terme. Celle-ci doit comprendre par ailleurs des recherches sur les points terminaux de pathogénicité et d'infectiosité.
Situations dans lesquelles les tests sont requis :
La toxicité à court terme du micro-organisme (vingt-huit jours au minimum) doit être décrite. Le choix du type de test doit être justifié et la durée de l'étude doit être décidée en fonction des données relatives à la toxicité aiguë et à l'élimination du micro-organisme. La meilleure voie d'administration doit être choisie sur l'avis des experts.
5.2.5.1 Effets sur la santé après exposition répétée par inhalation.
Les informations sur les effets sur la santé après exposition répétée par inhalation sont considérées comme nécessaires, particulièrement pour l'évaluation des risques sur le lieu de travail. L'exposition répétée pourrait affecter la capacité d'élimination de l'hôte (humain), notamment en renforçant la résistance du micro-organisme. En outre, pour une bonne évaluation des risques, il convient d'étudier la toxicité après exposition répétée aux contaminants, au milieu de culture, aux adjuvants et au micro-organisme, sans oublier que les adjuvants contenus dans le produit phytopharmaceutique peuvent influer sur la toxicité et l'infectiosité d'un micro-organisme.
Situations dans lesquelles le test est requis :
Des informations sur l'infectiosité, la pathogénicité et la toxicité à court terme du micro-organisme (voie respiratoire) sont exigées, à moins que les informations déjà fournies ne suffisent pour évaluer les effets sur la santé des personnes. Cela peut être le cas s'il est démontré que la substance testée ne comporte aucune fraction inhalable et/ou qu'aucune exposition répétée n'est envisagée.
5.2.6. Traitement proposé : premiers soins et traitements médicaux.
Les premiers soins à appliquer en cas d'infection ou de contamination des yeux doivent être prévus.
Les régimes thérapeutiques à mettre en oeuvre en cas d'ingestion ou de contamination des yeux ou de la peau doivent faire l'objet d'une description exhaustive. Il y a lieu de fournir, le cas échéant, toute information éventuellement disponible relative à l'efficacité des régimes thérapeutiques de substitution, fondée sur l'expérience pratique ou, à défaut, sur des considérations théoriques.
Des informations sur la résistance aux antibiotiques doivent également être fournies.
(Fin de la phase I).
Phase II.
5.3. Etudes spécifiques de toxicité, de pathogénicité et d'infectiosité.
Dans certains cas, il peut être nécessaire d'effectuer des études complémentaires pour clarifier les effets nocifs sur les personnes.
Des études de toxicité, de pathogénicité et d'infectiosité chroniques ainsi que de cancérogénicité et de toxicité reproductrice doivent notamment être effectuées lorsque les résultats des études précédentes indiquent que le micro-organisme peut avoir des effets à long terme sur la santé. Dans les cas où il y a production d'une toxine, il convient en outre d'effectuer des études cinétiques.
Les études requises peuvent être conçues sur une base individuelle, compte tenu des paramètres spécifiques à examiner et des objectifs à atteindre. Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
5.4. Etudes in vivo sur cellules somatiques.
Situations dans lesquelles les tests sont requis :
Si les résultats des études in vitro sont tous négatifs, des tests supplémentaires doivent être entrepris sur la base des autres informations utiles disponibles. Il peut s'agir d'une étude in vivo ou in vitro utilisant un système métabolique différent de celui ou de ceux précédemment employés.
Si le test cytogénétique in vitro est positif, il convient d'effectuer un essai in vivo sur des cellules somatiques (analyse des métaphases des cellules de la moelle osseuse de rongeur ou test du micronoyau chez les rongeurs).
Si l'un ou l'autre des tests de mutation génique in vitro est positif, il convient d'effectuer soit un test in vivo afin d'analyser la synthèse non programmée d'ADN, soit un "spot test" chez la souris.
5.5. Génotoxicité, études in vivo sur cellules germinales.
Objet et modalités du test :
Voir le point 5.4.
Situations dans lesquelles le test est requis :
Si l'un quelconque des résultats des tests effectués in vitro sur des cellules somatiques est positif, la réalisation d'un test in vivo pour déterminer les effets sur les cellules germinales peut se justifier. La nécessité d'effectuer ces tests doit être examinée cas par cas compte tenu des autres informations disponibles, relatives notamment aux modalités d'utilisation et aux situations prévisibles d'exposition. Des tests appropriés (tels que l'essai de létalité dominante) devront permettre d'examiner l'interaction avec l'ADN, de déterminer la possibilité de développer des effets héréditaires et, si possible, d'en réaliser une estimation quantitative. Compte tenu de la complexité de ce type d'études, il est reconnu que le recours à des études quantitatives suppose une justification solide.
(Fin de la phase II).
5.6. Synthèse : toxicité, pathogénicité et infectiosité pour les mammifères et évaluation globale.
Une synthèse de toutes les données et informations fournies en application des points 5.1 à 5.5 doit être présentée ; elle doit comporter une évaluation détaillée et critique desdites données sur la base de critères et de lignes directrices pertinents concernant l'évaluation et la prise de décision, compte tenu particulièrement des risques potentiels ou effectifs pour les êtres humains et les animaux ainsi que de l'ampleur, de la qualité et de la fiabilité de la base de données.
Il convient d'expliquer si l'exposition des animaux ou des êtres humains a des implications pour la vaccination ou le contrôle sérologique.
6. Résidus dans ou sur les produits traités, les denrées alimentaires et les aliments pour animaux.
(Arrêté du 17 décembre 2002, article 1er)
Introduction.
i) Les informations fournies, associées à celles présentées pour une ou plusieurs préparations contenant le micro-organisme, doivent être suffisantes pour réaliser une évaluation des risques pour les êtres humains et/ou les animaux d'une exposition au micro-organisme comme aux résidus et métabolites (toxines) qu'il laisse dans ou sur les plantes ou produits phytopharmaceutiques.
ii) En outre, les informations fournies doivent être suffisantes pour :
- arrêter une décision quant à l'inclusion éventuelle du micro-organisme dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- spécifier les conditions ou restrictions appropriées devant accompagner toute inclusion dans l'annexe 1 de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- le cas échéant, fixer les niveaux maximaux de résidus, les délais avant récolte destinés à protéger les consommateurs et les périodes d'attente destinées à protéger les professionnels amenés à manipuler les récoltes et les produits traités.
iii) Pour l'évaluation des risques liés aux résidus, les données expérimentales concernant les niveaux d'exposition aux résidus ne sont pas systématiquement exigées dès lors qu'il peut être démontré que le micro-organisme et ses métabolites ne sont pas dangereux pour les personnes humaines aux concentrations prévues pour l'utilisation autorisée. Les éléments de démonstration correspondants peuvent être fondés sur des sources bibliographiques publiques, l'expérience pratique et les informations visées aux sections 1, 2, 3 et 5.
6.1. Persistance et probabilité de multiplication dans ou sur les cultures, les aliments pour animaux ou les denrées alimentaires.
Il convient de fournir une estimation dûment justifiée de la persistance/compétitivité du micro-organisme et des métabolites secondaires sensibles (spécialement les toxines) dans ou sur les cultures, dans les conditions environnementales habituelles au moment de l'utilisation prévue et après celle-ci, en tenant compte notamment des informations présentées à la section 2.
En outre, le dossier de demande doit préciser dans quelle mesure et sur quelle base on estime que le micro-organisme possède (ou non) la faculté de se multiplier dans ou sur les végétaux ou produits végétaux ou lors des opérations de transformation des produits crus.
6.2. Informations complémentaires requises.
Les consommateurs peuvent être exposés aux micro-organismes pendant un temps considérable à la suite de la consommation de denrées alimentaires traitées. Il convient donc d'établir les effets potentiels sur les consommateurs sur la base d'études de chronicité ou de semi-chronicité visant à définir, aux fins de la gestion des risques, un seuil toxicologique (DJA, par exemple).
6.2.1. Résidus non viables.
On entend par micro-organisme non viable un micro-organisme incapable de se reproduire ou de transférer du matériel génétique.
Si la section 2, points 2.4 et 2.5, révèle le caractère persistant de quantités sensibles du micro-organisme ou de métabolites produits par celui-ci, spécialement des toxines, il y a lieu de fournir un relevé exhaustif des données expérimentales sur les résidus visées à l'annexe I, partie A, section 6, dès lors que le micro-organisme et/ou ses toxines sont susceptibles de se trouver sur ou dans les denrées ou aliments traités à des concentrations supérieures aux niveaux observés en conditions naturelles ou dans un état phénotypique différent.
Les conclusions relatives à la différence entre les concentrations naturelles et les concentrations élevées dues au traitement par le micro-organisme doivent se fonder sur des données obtenues par la voie expérimentale, et non sur des extrapolations ou calculs effectués à partir de modèles.
Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
6.2.2. Résidus viables.
Si les informations fournies en application du point 6.1 suggèrent qu'il y a persistance d'une quantité sensible de micro-organisme sur ou dans les produits, denrées ou aliments pour animaux traités, il convient d'en étudier les effets possibles sur les êtres humains et/ou sur les animaux, sauf s'il est démontré au titre de la section 5 que le micro-organisme et ses métabolites et/ou les produits issus de leur dégradation ne présentent pas de risque pour les êtres humains dans l'état et aux concentrations correspondant à l'utilisation autorisée.
Les conclusions relatives à la différence entre les concentrations naturelles et les concentrations élevées dues au traitement par le micro-organisme doivent se fonder sur des données obtenues par la voie expérimentale, et non sur des extrapolations ou calculs effectués à partir de modèles.
La persistance de résidus viables doit faire l'objet d'une attention particulière si les informations portées au titre des sections 2.3, 2.5 ou 5 relèvent une infectiosité ou une pathogénicité touchant les mammifères et/ou si toute autre information suggère un risque pour les consommateurs et/ou les professionnels. Dans ce cas, les autorités compétentes peuvent exiger des études semblables à celles prévues dans la partie A.
Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
6.3. Résumé et évaluation du comportement des résidus, sur la base des données fournies conformément aux points 6.1 et 6.2.
7. Devenir et comportement dans l'environnement
(Arrêté du 17 décembre 2002, article 1er)
Introduction.
i) Les informations sur l'origine, les propriétés et la survie du micro-organisme et de ses métabolites résiduels ainsi que sur l'utilisation proposée du micro-organisme forment la base de l'évaluation de son devenir et de son comportement dans l'environnement.
Des données expérimentales sont normalement exigées, à moins qu'il puisse être démontré que cette évaluation est réalisable à partir des informations déjà disponibles. Les éléments de démonstration correspondants peuvent être fondés sur des sources bibliographiques publiques, l'expérience pratique et les informations visées aux sections 1 à 6. On s'intéressera particulièrement à la fonction du micro-organisme dans les processus environnementaux (tels que définis à la section 2, point 2.1.2).
ii) Les informations fournies, associées aux autres informations pertinentes, et notamment à celles qui concernent une ou plusieurs préparations contenant le micro-organisme, doivent être suffisantes pour évaluer le devenir et le comportement du micro-organisme, de ses traces résiduelles et de ses toxines dès lors qu'ils présentent un risque pour la santé humaine et/ou pour l'environnement.
iii) En particulier, les informations fournies doivent être suffisantes pour :
- décider si le micro-organisme peut ou non être inclus dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- spécifier les conditions ou restrictions appropriées devant accompagner toute inclusion dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- définir les symboles de danger (une fois introduits), les mises en garde et les mentions types relatives à la nature des risques et aux conseils de prudence pour la protection de l'environnement à faire figurer sur les emballages (conteneurs) ;
- prévoir la dispersion, le devenir et le comportement dans l'environnement du micro-organisme et de ses métabolites ainsi que les durées correspondantes ;
- identifier les mesures nécessaires pour limiter au minimum la contamination de l'environnement et les incidences sur les espèces non visées.
iv) Tout métabolite sensible (c'est-à-dire qui présente un risque pour la santé humaine et/ou l'environnement) produit par l'organisme testé dans toutes les conditions environnementales appropriées doit faire l'objet d'une caractérisation. Dans les cas où des métabolites sensibles sont présents au sein du micro-organisme, ou produits par ce dernier, les données prévues à l'annexe I, partie A, point 7, peuvent être exigées dès lors que toutes les conditions ci-après sont réunies :
- le métabolite sensible est stable hors du micro-organisme (voir point 2.8) ;
- l'effet toxique du métabolite est indépendant de la présence du micro-organisme ;
- on prévoit que le métabolite sensible se retrouve dans l'environnement à des concentrations considérablement plus élevées que dans les conditions naturelles.
v) Les informations disponibles sur les liens avec des souches sauvages apparentées présentes dans la nature doivent être prises en compte.
vi) Avant d'engager les études visées ci-après, il appartient au demandeur d'obtenir l'accord des autorités compétentes sur l'opportunité de mener de telles études et, dans l'affirmative, sur le type d'études à entreprendre. Les informations visées dans les autres sections doivent également être prises en considération.
7.1. Persistance et multiplication.
Il convient de fournir, le cas échéant, des informations pertinentes sur la persistance et la multiplication du micro-organisme dans tous les milieux environnementaux, en accordant une attention particulière :
- à la compétitivité dans les conditions environnementales normales au moment de l'utilisation proposée et après celle-ci ;
- à la dynamique de population sous des climats marqués par des extrêmes à caractère saisonnier ou régional (étés particulièrement chauds, hivers particulièrement froids, précipitations abondantes) et aux pratiques agricoles mises en oeuvre après l'application du produit.
Il convient d'indiquer les niveaux estimatifs de présence du micro-organisme sur une période donnée après utilisation du produit dans les conditions proposées.
7.1.1. Sols.
Les informations sur la viabilité/la dynamique de population doivent être présentées pour plusieurs types de sols cultivés ou non cultivés caractéristiques des différentes régions de la Communauté où l'utilisation du produit est prévue ou déjà effective. Il convient à cet effet d'observer les dispositions prévues dans la partie A, point 7.1, introduction, en ce qui concerne le choix et le mode de prélèvement des sols. S'il est prévu d'utiliser l'organisme testé en association avec d'autres constituants tels que la laine de roche, ceux-ci doivent être inclus dans la batterie de tests.
7.1.2. Eau.
Des informations doivent être fournies sur la viabilité/la dynamique de population du micro-organisme dans les systèmes sédimentaires/hydrauliques, tant dans l'obscurité qu'en pleine lumière.
7.1.3. Air.
En cas de préoccupations particulières liées à l'exposition des opérateurs, des ouvriers ou de toute autre personne présente, des informations sur les concentrations dans l'air peuvent être nécessaires.
7.2. Mobilité.
La propagation éventuelle du micro-organisme et des produits issus de sa dégradation dans tous les milieux environnementaux doit faire l'objet d'une évaluation, sauf s'il peut être démontré que toute exposition des différents milieux considérés au micro-organisme est improbable.
Dans cette perspective, on s'intéressera particulièrement à l'utilisation prévue (dans les champs ou sous serre, en application sur les sols ou sur les cultures), au cycle de vie du micro-organisme et à ses différents stades, à la présence de vecteurs, à la persistance et à la capacité du micro-organisme à coloniser des habitats adjacents.
La propagation, la persistance et les distances probables de dissémination appellent une attention particulière si une toxicité, une infectiosité ou une pathogénicité ont été rapportées ou si d'autres informations suggèrent la possibilité de risques pour les personnes, les animaux ou l'environnement. Dans ce cas, les autorités compétentes peuvent exiger des études semblables à celles prévues dans la partie A. Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
8. Effets sur les organismes non visés
(Arrêté du 17 décembre 2002, article 1er)
Introduction.
i) Les informations concernant l'identité et les propriétés biologiques ainsi que les informations complémentaires visées aux sections 1 à 3 et 7 sont capitales pour évaluer les effets sur les espèces non visées. En complément, des informations utiles sur, d'une part, le devenir et le comportement du micro-organisme dans l'environnement et, d'autre part, les niveaux de résidus présents dans les végétaux figurent, respectivement, aux sections 7 et 6. Associées aux renseignements concernant la nature de la préparation et son mode d'utilisation, elles permettent de définir la nature de l'exposition potentielle et d'en délimiter l'étendue. Les informations fournies au titre de la section 5 fournissent des informations essentielles en ce qui concerne les effets sur les mammifères et les mécanismes en jeu. Des données expérimentales sont normalement exigées, à moins qu'il ne puisse être démontré que l'évaluation des effets sur les organismes non visés est réalisable à partir des informations déjà disponibles.
ii) La sélection des organismes non visés à inclure dans l'étude des effets sur l'environnement doit être fondée sur la nature du micro-organisme (et notamment la spécificité d'hôte, le mode d'action et le mode de fonctionnement écologique de l'organisme). Ces éléments doivent permettre de choisir en vue des tests les organismes appropriés, à savoir, par exemple, des organismes étroitement apparentés à l'organisme cible.
iii) Les informations fournies, jointes à celles qui concernent une ou plusieurs préparations contenant le micro-organisme, doivent être suffisantes pour permettre d'évaluer les effets sur les espèces non visées (flore et faune) dont l'exposition au micro-organisme peut être dangereuse, lorsqu'elles ont une importance pour l'environnement. Ces effets peuvent être dus à une exposition unique, prolongée ou répétée et peuvent être réversibles ou irréversibles.
iv) En particulier, les informations fournies sur le micro-organisme, associées aux autres informations pertinentes et aux informations relatives à une ou plusieurs préparations contenant le micro-organisme, doivent être suffisantes pour :
- décider si le micro-organisme peut ou non être inclus dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- spécifier les conditions ou restrictions appropriées devant accompagner toute inclusion dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques ;
- permettre une évaluation des risques à court terme comme à long terme pour les espèces non visées (populations, communautés et processus, selon le cas) ;
- classer le micro-organisme selon les risques biologiques qu'il présente ;
- préciser les précautions à prendre pour protéger les espèces non visées ; et
- définir les symboles de danger (une fois introduits), les mises en garde et les mentions types relatives à la nature des risques et aux conseils de prudence pour la protection de l'environnement à faire figurer sur les emballages (conteneurs).
v) Il y a lieu de mentionner tous les effets potentiellement néfastes constatés au cours des investigations de routine sur les effets environnementaux. Il convient également, sur demande des autorités compétentes, d'effectuer et de relater les études supplémentaires qui se révéleraient nécessaires pour identifier les mécanismes susceptibles d'être en cause et évaluer l'importance des effets constatés. Il est indispensable de rapporter toutes les données et informations biologiques disponibles concourant à l'évaluation du profil écologique du micro-organisme.
vi) Pour toutes les études, la dose moyenne employée, exprimée en unités colonisatrices par kilogramme de poids corporel ainsi que dans d'autres unités appropriées, doit être mentionnée.
vii) Des études séparées sur les métabolites sensibles (et notamment les toxines) peuvent s'imposer lorsque ces produits peuvent présenter un risque non négligeable pour les organismes non visés et que les résultats des études concernant le micro-organisme ne permettent pas d'évaluer leurs effets. Avant d'engager les travaux, il appartient au demandeur d'obtenir l'accord des autorités compétentes sur l'opportunité de mener de telles études et, dans l'affirmative, sur le type d'études à entreprendre. Les informations visées aux sections 5, 6 et 7 doivent être prises en considération.
viii) Pour faciliter l'évaluation des résultats obtenus et de leur portée, il y a lieu, dans la mesure du possible, d'utiliser pour les différents tests la même souche (ou origine certifiée) de chacune des espèces concernées.
ix) Les tests sont obligatoires, sauf s'il peut être démontré que l'organisme non visé ne sera pas exposé au micro-organisme. S'il est démontré que le micro-organisme n'a aucun effet toxique, pathogène ou infectieux sur les vertébrés ou les végétaux, les investigations se limitent aux réactions des organismes non visés appropriés.
8.1. Effets sur les oiseaux.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les oiseaux.
8.2. Effets sur les organismes aquatiques.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les organismes aquatiques.
8.2.1. Effets sur les poissons.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les poissons.
8.2.2. Effets sur les invertébrés d'eau douce.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les invertébrés d'eau douce.
8.2.3. Effets sur la croissance des algues.
Objet du test :
Des informations doivent être fournies en ce qui concerne les effets sur la croissance des algues, leur taux de croissance et leur capacité de récupération.
8.2.4. Effets sur les végétaux autres que les algues.
Objet du test :
Des informations doivent être fournies en ce qui concerne les effets sur les végétaux autres que les algues.
8.3. Effets sur les abeilles.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les abeilles.
8.4. Effets sur les arthropodes autres que les abeilles.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les arthropodes autres que les abeilles. Les espèces à inclure dans le test doivent être sélectionnées sur la base des usages potentiels des produits phytopharmaceutiques concernés (application foliaire ou sur les sols, par exemple). Il convient d'accorder une attention particulière aux organismes utilisés aux fins de lutte biologique et à ceux qui jouent un rôle important dans le cadre d'un système intégré de lutte contre les nuisibles.
8.5. Effets sur les vers de terre.
Objet du test :
Des informations doivent être fournies sur la toxicité, l'infectiosité et la pathogénicité pour les vers de terre.
8.6. Effets sur les micro-organismes non visés présents dans les sols.
Les effets sur les micro-organismes non visés et sur leurs prédateurs (par exemple, des protozoaires, dans le cas des inoculants bactériens) doivent être signalés. Un avis d'expert est exigé pour décider s'il convient d'engager des études supplémentaires.
Cette décision doit prendre en considération les informations disponibles au titre de la présente section, mais également d'autres sections, et notamment les données relatives à la spécificité du micro-organisme et aux situations d'exposition prévues. Les observations réalisées lors de tests d'efficacité peuvent également fournir à cet égard des informations utiles.
Une attention particulière doit être accordée aux organismes utilisés dans le cadre de la gestion intégrée des cultures.
8.7. Etudes supplémentaires.
Les études supplémentaires peuvent comprendre d'autres études pointues sur des espèces ou des systèmes supplémentaires (tels qu'un réseau d'égouts) ou des études à un niveau supérieur consacrées, par exemple, aux effets chroniques ou sublétaux sur certains organismes non visés ou encore aux effets sur leur reproduction.
Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
9. Synthèse et évaluation des incidences sur l'environnement.
(Arrêté du 17 décembre 2002, article 1er)
Il convient d'élaborer une synthèse et une évaluation de toutes les données concernant l'incidence sur l'environnement. Le document doit comprendre une évaluation critique et détaillée de ces données dans la perspective des lignes directrices et des critères importants pour l'évaluation et la prise de décision, en prêtant une attention particulière aux risques éventuels ou effectifs pour l'environnement et les espèces non visées ainsi qu'à l'importance, à la qualité et à la fiabilité de la base de données. Doivent notamment être traités :
- la dissémination et le devenir dans l'environnement, avec mention des durées correspondantes ;
- l'identification des espèces et des populations non visées susceptibles d'être affectées ainsi que l'ampleur de leur exposition potentielle ;
- l'identification des précautions nécessaires pour éviter ou réduire au minimum la contamination de l'environnement et protéger les espèces non visées.
Annexe II : Conditions à remplir pour introduire le dossier d'autorisation d'un produit phytopharmaceutique
Modifié par Arrêté 1996-02-01 art. 3, art. 4 JORF 20 février 1996 et Arrêté 2002-12-17 art. 2 JORF 14 janvier 2003
Introduction.
L'information doit :
1.1. Comprendre un dossier technique fournissant les informations nécessaires pour évaluer l'efficacité et les risques prévisibles, immédiats ou à plus long terme, que le produit phytopharmaceutique peut comporter pour l'homme, l'animal et l'environnement, contenant au moins les résultats des études visées ci-après.
1.2. Le cas échéant, être recueillie conformément à la version la plus récente des lignes directrices, visées ou décrites dans la présente annexe ; pour les études commencées avant la mise en vigueur de la modification de la présente annexe, l'information doit être recueillie conformément à des lignes directrices adéquates, validées à l'échelon national ou international, ou, en leur absence, à des lignes directrices acceptées par l'autorité compétente.
1.3. Comprendre, si la ligne directrice ne convient pas ou n'est pas décrite, ou si l'on a utilisé une autre ligne directrice que celles qui sont visées dans la présente annexe, une justification de la ligne directrice utilisée qui soit acceptable pour l'autorité compétente.
En particulier lorsqu'il est fait référence dans cette annexe à une méthode CE qui est la transposition d'une méthode mise au point par une organisation internationale (par exemple l'O.C.D.E.), les Etats membres peuvent accepter que l'information requise soit recueillie conformément à la version la plus récente de cette méthode si, au début des études, la méthode CE n'a pas encore été mise à jour.
1.4. Comprendre, si l'autorité compétente l'exige, une description complète des lignes directrices utilisées, à moins qu'il n'y soit fait référence ou qu'elles soient décrites dans la présente annexe, ainsi qu'une description complète de toute variante ainsi que sa justification, acceptable pour l'autorité compétente.
1.5. Comprendre un rapport complet et impartial des études menées ainsi que leur description complète ou une justification acceptable pour l'autorité compétente :
- lorsque certaines données ou informations particulières qui ne semblent pas nécessaires en raison de la nature de la substance ou des utilisations qui en sont proposées ne sont pas fournies,
ou
- lorsqu'il n'est pas scientifiquement nécessaire ou techniquement possible de fournir les informations et les données.
1.6. Le cas échéant, avoir été recueillie conformément aux dispositions de la directive n° 86/609/C.E.E.
2.1. Les essais et analyses doivent être effectués conformément aux principes fixés dans la directive n° 87/18/C.E.E. lorsqu'ils ont pour but de recueillir des données sur les propriétés intéressant la santé humaine et animale ou l'environnement et/ou sur la sécurité dans ces domaines.
2.2. Les essais et analyses exécutés conformément aux dispositions de la section 6, points 6.2 à 6.7, de la présente annexe sont réalisés par des services ou organismes d'essais officiels ou officiellement reconnus, remplissant au moins les conditions suivantes :
- avoir à leur disposition un personnel scientifique et technique suffisant, ayant l'instruction, la formation, les connaissances et expériences techniques nécessaires pour assumer les fonctions qui leur sont assignées ;
- avoir à leur disposition l'équipement approprié nécessaire pour une exécution correcte des essais et des mesures qu'ils prétendent être en mesure de réaliser. Cet équipement doit être correctement entretenu et calibré, le cas échéant, avant et après sa mise en service conformément à un programme établi ;
- avoir à leur disposition des champs d'essais appropriés et, si nécessaire, des serres, des chambres de croissance ou des locaux de stockage. L'environnement dans lequel les essais sont réalisés ne doit pas fausser leurs résultats ou nuire à la précision demandée de la mesure ;
- mettre à la disposition de tout le personnel compétent les modes opératoires et protocoles pour les essais ;
- fournir, si l'autorité compétente le demande, avant le commencement d'un essai, des informations détaillées sur cet essai, en indiquant au moins le lieu de l'essai et les produits qu'il concerne ;
- faire en sorte que la qualité des travaux exécutés soit adaptée au type, à la gamme, au volume et à l'objectif de ces travaux ;
- conserver l'enregistrement de l'ensemble des observations initiales, calculs et données dérivées, ainsi que les enregistrements relatifs à l'étalonnage et le rapport final aussi longtemps que le produit concerné est autorisé dans la Communauté.
2.3. Les Etats membres exigent des services et organismes d'essais officiellement reconnus et, s'il y lieu, des services et organismes officiels qu'ils :
- communiquent à l'autorité nationale compétente l'ensemble des informations détaillées nécessaires pour prouver qu'ils sont à même de remplir les conditions prévues au point 2.2 ;
- acceptent à tout moment les inspections que chaque Etat membre organise régulièrement sur son territoire afin de vérifier la conformité avec les conditions prévues au point 2.2.
2.4. Par dérogation au point 2.1, les Etats membres peuvent également appliquer les dispositions des points 2.2 et 2.3 aux essais et aux analyses effectués sur leur territoire, visant à recueillir des données sur les propriétés et/ou la sécurité des préparations en ce qui concerne les abeilles et les arthropodes utiles autres que les abeilles, et qui ont effectivement débuté au plus tard le 31 décembre 1999.
2.5. Par dérogation au point 2.1, les Etats membres peuvent également appliquer les dispositions des points 2.2 et 2.3 aux essais contrôlés sur les résidus, effectués sur leur territoire conformément aux dispositions de la section 8 "Résidus dans ou sur les produits, la nourriture et l'alimentation traités" avec des produits phytosanitaires contenant des substances actives déjà présentes sur le marché au 25 juillet 1993 lorsqu'ils ont effectivement débuté au plus tard le 31 décembre 1997.
2.6. Par dérogation au point 2.1, pour les substances actives constituées de micro-organismes ou de virus, les tests et analyses effectués afin de recueillir des données sur les propriétés et/ou la sécurité en ce qui concerne des aspects autres que la santé humaine peuvent être réalisés par des services ou organismes d'expérimentation officiels ou officiellement reconnus remplissant au minimum les conditions visées aux points 2.2 et 2.3 de l'introduction de l'annexe II.
Partie A : Préparations chimiques.
Article Annexe II
Modifié par JORF 20 février 1996
1. Identité du produit phytopharmaceutique
(Arrêté du 1er février 1996, article 2)
Les informations fournies, considérées avec celles concernant la (les) substance(s) active(s), doivent être suffisantes pour permettre d'identifier précisément les préparations et de les définir quant à leur spécification et nature. Les informations et données mentionnées, sauf spécification contraire, sont nécessaires pour tous les produits phytosanitaires.
1.1. Demandeur (nom, adresse, etc.).
Le nom et l'adresse du demandeur (adresse permanente dans la Communauté) doivent être donnés, tout comme le nom, la qualité, les numéros de téléphone et de télécopieur de la personne à contacter.
Si, en outre, le demandeur dispose d'un bureau, d'un agent ou d'un représentant dans l'Etat membre dans lequel l'autorisation est demandée, le nom et l'adresse du bureau, de l'agent ou du représentant local doivent être fournis avec le nom, la qualité, les numéros de téléphone et de télécopieur de la personne à contacter.
1.2. Fabricant du produit phytopharmaceutique et de la (des) substance(s) active(s) (nom, adresse, etc.), y compris l'emplacement des installations.
Le nom et l'adresse du fabricant de la préparation et de chaque substance active contenue dans la préparation doivent être fournis tout comme le nom et l'adresse de chaque installation dans laquelle la préparation et la substance active sont préparées. Un point de contact (de préférence un point de contact central avec nom, numéros de téléphone et de télécopie) doit être prévu dans chaque cas.
Si la substance active provient d'un fabricant pour lequel les données visées à l'annexe I n'ont pas été fournies auparavant, il convient de préciser la pureté et de fournir les informations détaillées requises à l'annexe I concernant les impuretés.
1.3. Nom commercial ou nom commercial proposé et, le cas échéant, le numéro de code de développement attribué au fabricant pour la préparation.
Tous les noms commerciaux, anciens et courants, noms commerciaux proposés et numéros de code de développement de la préparation ainsi que les noms et numéros courants doivent être fournis. Si les noms commerciaux et numéros de code mentionnés s'appliquent à des préparations similaires, mais différentes (éventuellement hors d'usage), fournir une information complète concernant ces différences. (Le nom commercial proposé ne doit pas donner lieu à confusion avec le nom commercial des produits phytopharmaceutiques déjà déposés).
1.4. Informations détaillées d'ordre quantitatif et qualitatif sur la composition de la préparation (substance"s" active"s" et autres produits).
1.4.1. Donner les informations suivantes pour les préparations :
- la concentration de la (des) substance(s) active(s) technique(s) et de la (des) substance(s) active(s) pure(s) ;
- la concentration des autres produits.
Les concentrations doivent être exprimées dans les termes prévus à l'article 6, paragraphe 2, de la directive 78/631/CEE.
1.4.2. Pour les substances actives, indiquer leur nom commun ISO ou leur nom commun ISO proposé ainsi que leur numéro CIPAC et, s'il existe, leur numéro CEE (Einecs ou Elincs). Le cas échéant, indiquer le sel, l'ester, l'anion ou le cation présent.
1.4.3. Si possible, identifier les autres produits de la formule par leur nom chimique précisé à l'annexe I de la directive 67/548/CEE ou, si tel n'est pas le cas, selon les nomenclatures de l'UICPA et des CA. Indiquer leur structure ou formule développée de structure. Pour chaque composant des autres produits de la formule, indiquer, s'ils existent, le numéro CEE (Einecs ou Elincs) et le numéro CAS. Si l'information fournie ne permet pas d'identifier exactement les produits considérés de la formule, fournir une spécification appropriée. S'il existe, fournir aussi le nom commercial de ces produits.
1.4.4. Indiquer la fonction des produits compris dans la formule :
- adhésif ;
- agent antimoussant ;
- antigel ;
- liant ;
- tampon ;
- agent porteur ;
- déodorant ;
- agent dispersant ;
- teinture ;
- émétique ;
- émulsifiant ;
- fertilisant ;
- conservateur ;
- agent odorant ;
- parfum ;
- agent d'appétence ;
- répulsif ;
- phytoprotecteur ;
- solvant ;
- stabilisant ;
- synergiste ;
- épaississant ;
- agent mouillant ;
- divers (à spécifier).
1.5. Etat physique et nature de la préparation (concentré émulsionnable, poudre mouillable, solution, etc.).
1.5.1. Le type et le code de la préparation doivent être spécifiés conformément au "Catalogue des types de formulation de pesticides et système de code international (Monographie technique GIFAP n° 2, 1989)".
Si une préparation spécifique n'est pas définie précisément dans la présente publication, donner une description complète de la nature physique et de l'état de la préparation en même temps qu'une proposition de description convenable du type de préparation et une proposition de définition y relative.
1.6. Fonction (herbicide, insecticide, etc.).
Préciser la fonction à retenir parmi les suivantes :
- acaricide ;
- bactéricide ;
- fongicide ;
- herbicide ;
- insecticide ;
- molluscicide ;
- nématicide ;
- phéromone ;
- régulateur de croissance ;
- répulsif ;
- rodenticide ;
- médiateurs chimiques ;
- taupicide ;
- virucide ;
- autres (à préciser).
Article Annexe II En savoir plus sur cet article...
Modifié par Arrêté 1996-02-01 art. 2 JORF 20 février 1996
2. Propriétés physiques, chimiques et techniques du produit phytopharmaceutique
(Arrêté du 1er février 1996, article 2)
Indiquer dans quelle mesure les produits phytopharmaceutiques pour lesquels l'autorisation est demandée sont conformes aux spécifications FAO pertinentes, établies par le Groupe des spécifications relatives aux pesticides, de la liste FAO d'experts des spécifications, critères d'homologation et normes d'application des pesticides. Donner une description détaillée et justifier les différences par rapport aux spécifications FAO.
2.1. Aspect (couleur et odeur).
Spécifier éventuellement la couleur et l'odeur ainsi que l'état physique de la préparation.
2.2. Propriétés explosives et oxydantes.
2.2.1. Les propriétés explosives des préparations doivent être relatées conformément à la méthode CEE A 14. Si des informations thermodynamiques disponibles indiquent avec un degré de certitude suffisant que la préparation ne peut produire de réaction exothermique, ces informations suffisent à prouver qu'il n'est pas nécessaire de déterminer les propriétés explosives de la préparation.
2.2.2. Déterminer et indiquer conformément à la méthode CEE A 17 les propriétés oxydantes des préparations qui se présentent sous forme de solides. Pour d'autres préparations, justifier la méthode utilisée. Il est inutile de déterminer les propriétés oxydantes s'il peut être démontré avec un degré de certitude suffisant sur la base des informations thermodynamiques que la préparation ne peut produire de réactions exothermiques avec des matériaux combustibles.
2.3. Point d'éclair et autres indications sur l'inflammabilité ou l'ignition spontanée.
Déterminer et indiquer conformément à la méthode CEE A 9 le point d'éclair des liquides contenant des solvants inflammables. Déterminer l'inflammabilité des préparations solides et des gaz et les indiquer selon les méthodes CEE A 10, A 11 et A 12. Déterminer l'auto-inflammabilité des préparations et les indiquer selon les méthodes CEE A 15 ou A 16 selon le cas et/ou, si nécessaire, selon l'essai en cage de Bowes-Cameron des Nations unies (recommandations des Nations unies relatives au transport des marchandises dangereuses, chapitre 14, point 14.3.4).
2.4. Acidité/alcalinité et, si nécessaire, valeur du pH.
2.4.1. Dans le cas de préparations acides (pH 4) ou alcalines (pH 10), déterminer et indiquer l'acidité ou l'alcalinité ainsi que la valeur du pH selon les méthodes CIPAC MT 31 et MT 75 respectivement.
2.4.2. Le cas échéant (si elle doit être utilisée sous forme de dilution aqueuse), déterminer et indiquer le pH d'une dilution, émulsion ou dispersion aqueuse à 1 p. 100 de la préparation selon la méthode CIPAC MT 75.
2.5. Viscosité et tension superficielle.
2.5.1. Dans le cas de préparations liquides destinées à être appliquées à très bas volume (T.B.V.), déterminer et relater leur viscosité cinématique selon la ligne directrice n° 114 de l'O.C.D.E.
2.5.2. Pour les liquides non newtoniens, déterminer et indiquer leur viscosité en même temps que les conditions d'essai.
2.5.3. Pour les préparations liquides, déterminer et indiquer les tensions superficielles selon la méthode CEE A 5.
2.6. Densité relative et densité globale.
2.6.1. Déterminer et indiquer la densité des préparations liquides selon la méthode CEE A 3.
2.6.2. Déterminer et indiquer la masse volumique (après tassement) en vrac des préparations se présentant sous forme de poudre ou de granules selon les méthodes CIPAC MT 33, MT 159 ou MT 169, selon le cas.
2.7. Stabilité pendant le stockage - Stabilité et durée de conservation. Incidence de la lumière, de la température et de l'humidité sur les caractéristiques techniques du produit phytopharmaceutique.
2.7.1. Déterminer et indiquer la stabilité de la préparation après stockage à 54 °C pendant quatorze jours selon la méthode CIPAC MT 46.
Il peut être nécessaire de prévoir d'autres durées et/ou d'autres températures (par exemple huit semaines à 40 °C ou douze semaines à 35 °C ou dix-huit semaines à 30 °C) si la préparation est thermosensible.
Si la concentration des substances actives après le test de stabilité à la chaleur a diminué de plus de 5 p. 100 de la concentration constatée initialement, déclarer la concentration minimale et donner des informations sur les produits de dégradation.
2.7.2. En outre, pour les préparations liquides, déterminer et indiquer l'effet de faibles températures sur la stabilité selon les méthodes CIPAC MT 39, MT 48, MT 51 ou MT 54, selon le cas.
2.7.3. Indiquer la durée de conservation de la préparation à température ambiante. Si elle est inférieure à deux ans, indiquer cette durée en mois en donnant les spécifications de température appropriées. La monographie n° 17 du GIFAP contient des informations utiles.
2.8. Caractéristiques du produit phytosanitaire.
Déterminer les caractéristiques techniques de la préparation en vue d'une décision relative à son acceptabilité.
2.8.1. Mouillabilité.
Déterminer et indiquer la mouillabilité des préparations solides utilisées en dilution (poudres mouillables, poudres hydrosolubles, granulés hydrosolubles et granulés hydrodispersibles) selon la méthode CIPAC MT 53.3.
2.8.2. Formation d'une mousse persistante.
Déterminer et indiquer la persistance de mousse pour les préparations destinées à être diluées dans l'eau selon la méthode CIPAC MT 47.
2.8.3. Mise en suspension, tenue en suspension.
Déterminer et indiquer la faculté de passer en suspension de produits hydrodispersibles (par exemple poudres mouillables, granules hydrodispersibles, suspensions concentrées) selon la méthode CIPAC MT 15, MT 161 ou MT 168, selon le cas.
Pour les produits hydrodispersibles (suspensions concentrées et granules hydrodispersibles), déterminer et indiquer la spontanéité de la dispersion selon les méthodes CIPAC MT 160 ou MT 174, selon le cas.
2.8.4. Stabilité de la dilution.
Déterminer et indiquer la stabilité de la dilution de poudres hydrosolubles selon la méthode CIPAC MT 41.
2.8.5. Test du tamis humide, test du tamis sec.
Pour garantir que les poudres à poudrer aient une distribution appropriée de la taille de leurs particules, effectuer et indiquer une épreuve au tamis sec selon la méthode CIPAC MT 59.1.
S'il s'agit de produits hydrodispersibles, effectuer et indiquer une épreuve au tamis humide selon la méthode CIPAC MT 59.3 ou MT 167, selon le cas.
2.8.6. Distribution granulométrique (poudres pour poudrage et mouillables, granulés), teneur en poussières/particules fines (granulés), usure et friabilité (granulés).
2.8.6.1. S'il s'agit de poudres, déterminer et indiquer la distribution granulométrique des particules selon la méthode O.C.D.E. 110.
Indiquer la granulométrie nominale des granules destinés à une application directe, déterminée selon la méthode CIPAC MT 58.3 et des granules hydrodispersibles selon la méthode CIPAC MT 170.
2.8.6.2. Déterminer et indiquer la teneur en poussières des préparations granulées selon la méthode CIPAC MT 171. S'il convient de déterminer l'exposition de l'opérateur, déterminer et indiquer la taille des particules de poussière selon la méthode O.C.D.E. 110.
2.8.6.3. Déterminer et indiquer les caractéristiques de friabilité et d'usure des granulés dès que des méthodes internationalement convenues sont disponibles. Si des données sont déjà disponibles, elles doivent être précisées en même temps que la méthode utilisée.
2.8.7. Faculté d'émulsification, de réémulsification, stabilité de l'émulsion.
2.8.7.1. Déterminer et indiquer la faculté d'émulsification, la stabilité de l'émulsion et la faculté de réémulsification des préparations sous forme d'émulsions selon la méthode CIPAC MT 36 ou MT 173, selon le cas.
2.8.7.2. Déterminer et indiquer la stabilité des émulsions diluées et des préparations sous forme d'émulsions selon la méthode CIPAC MT 20 ou MT 173.
2.8.8. Faculté d'écoulement, de vidage (faculté de rinçage) et de transformation en poussières.
2.8.8.1. Déterminer et indiquer la faculté d'écoulement des préparations granulées selon la méthode CIPAC MT 172.
2.8.8.2. Déterminer et indiquer la faculté de vidage (y compris du résidu de rinçage) des suspensions (par exemple suspensions concentrées, suspo-émulsions) selon la méthode CIPAC MT 148.
2.8.8.3. Déterminer et indiquer la faculté de transformation en poussières des poudres pour poudrage après une opération de stockage accélérée de mise en stockage conformément au point 2.7.1, selon la méthode CIPAC MT 34 ou une autre méthode appropriée.
2.9. Compatibilité physique et chimique avec d'autres produits, y compris les produits phytopharmaceutiques avec lesquels son usage sera autorisé
2.9.1. Indiquer la compatibilité physique des mélanges en cuve sur la base de méthodes de testage interne. Un test pratique serait une méthode alternative acceptable.
2.9.2. Déterminer et indiquer la compatibilité chimique des mélanges en cuve, sauf si l'examen des propriétés particulières des préparations établissait avec un degré de certitude suffisant qu'aucune réaction ne peut avoir lieu. Dans de tels cas il suffit de donner cette information pour justifier qu'il n'est pas nécessaire de procéder à la détermination pratique de la compatibilité chimique.
2.10. Adhérence et répartition sur semences.
Dans le cas de préparations pour le traitement des semences, analyser et indiquer la distribution et l'adhérence, selon la méthode CIPAC MT 175 pour la distribution.
2.11. Résumé et évaluation des données indiquées aux points 2.1 à 2.10.
3. Données relatives à l'application
(Arrêté du 1er février 1996, article 2)
3.1. Domaine d'utilisation, par exemple champ, serre, stockage de produits végétaux, jardin.
Le(s) domaine(s) d'application, existant(s) et proposé(s), pour des préparations contenant la substance active doit (doivent) être spécifié(s) parmi les suivants :
- champs : utilisation en pleine terre comme en agriculture, horticulture, sylviculture, viticulture ;
- serre ;
- jardins publics ;
- désherbage des terres non cultivées ;
- jardins domestiques ;
- plantes d'intérieur ;
- stockage de produits végétaux ;
- autres (à préciser).
3.2. Effets sur les organismes nuisibles, par exemple poison par contact, par inhalation, poison stomacal, fongitoxique, etc., systémique ou non chez les végétaux.
Indiquer la nature des effets sur les organismes nuisibles :
- action par contact ;
- action sur l'estomac ;
- action par inhalation ;
- action fongitoxique ;
- action fongistatique ;
- déshydratant ;
- inhibiteur de reproduction ;
- autres (à préciser).
Spécifier si le produit est transloqué à l'intérieur des végétaux.
3.3. Modalités de l'utilisation envisagée, par exemple types d'organismes nuisibles combattus et/ou végétaux ou produits végétaux à protéger.
Fournir des précisions sur l'utilisation envisagée.
Le cas échéant, indiquer les effets obtenus, par exemple inhibition de la germination, retardement de la maturation, inhibition de la croissance de la tige, amélioration de la fertilité, etc.
3.4. Doses d'application.
Pour chaque méthode d'application et chaque usage, indiquer la dose d'application par unité traitée (ha, m2, m3), en grammes ou kilogrammes de préparation et de substance active.
Exprimer normalement les doses d'application en g ou kg/ha ou encore en kg/m3 et, si nécessaire en g ou kg/t ; pour les serres et les jardins domestiques, exprimer les doses d'utilisation en g ou kg/100 m2 ou g ou kg/m3.
3.5. Concentration de la substance active dans le support utilisé (par exemple dans le produit de pulvérisation dilué, les appâts ou les semences traitées).
Indiquer la teneur en substance active, selon le cas, en g/l ; g/kg ; mg/kg ou en g/t.
3.6. Méthode d'application.
Décrire in extenso la méthode d'application, en indiquant, le cas échéant, le type d'équipement à utiliser ainsi que le type et le volume du diluant à utiliser par unité de surface ou de volume.
3.7. Nombre et calendrier des applications et durée de la protection.
Indiquer le nombre maximal d'applications avec leur calendrier. Indiquer, le cas échéant, les stades de développement de la culture ou des végétaux ainsi que ceux des organismes nuisibles. Si possible, préciser l'intervalle en jours entre deux applications.
Indiquer la durée de protection assurée pour chaque application et pour le nombre maximal d'applications.
3.8. Périodes d'attente nécessaires ou autres précautions à prendre pour éviter des effets phytotoxiques sur les cultures ultérieures.
Le cas échéant, indiquer les périodes d'attente minimales entre la dernière application et le semis ou l'implantation des cultures suivantes, les périodes nécessaires pour prévenir des effets phytotoxiques sur les cultures suivantes, découlant des données figurant au point 6.6 de cette annexe.
Indiquer éventuellement les limitations quant au choix des cultures ultérieures.
3.9. Instructions d'emploi proposées.
Faire figurer les instructions proposées relatives à l'utilisation de la préparation sous forme imprimée sur les étiquettes et notices.
(Arrêté du 1er février 1996, article 2)
4.1. Emballage (type, matériaux, dimension, etc.), compatibilité de la préparation avec les matériaux d'emballage proposés.
4.1.1. Décrire de manière exhaustive l'emballage à utiliser et le spécifier quant aux matériaux utilisés, mode de fabrication (par exemple extrudé, soudé, etc.), sa dimension et sa capacité, mention de son ouverture, le type de fermeture et le scellement. Il doit être conçu conformément aux critères et lignes directrices spécifiés dans les "Directives de la FAO pour le conditionnement et le stockage des pesticides".
4.1.2. Déterminer et préciser l'adéquation de l'emballage, y compris les dispositifs de fermeture, sur le plan de la solidité, de l'imperméabilité et de la résistance à des conditions de transport et de manutention normales, selon les méthodes ADR 3552, 3553, 3560, 3554, 3555, 3556, 3558 ou les méthodes ADR appropriées pour conteneurs vrac intermédiaires et, si des fermetures inviolables pour les enfants sont nécessaires pour la préparation considérée, selon la norme ISO 8317.
4.1.3. Indiquer la résistance du matériau d'emballage au contenu selon la monographie GIFAP n° 17.
4.2. Méthodes de nettoyage de l'équipement utilisé pour l'application.
Décrire en détail les méthodes de nettoyage à utiliser pour l'équipement d'application et les vêtements de protection. Analyser et indiquer de manière exhaustive l'efficacité de la méthode de nettoyage.
4.3. Périodes de réintroduction, périodes d'attente nécessaires ou autres précautions à prendre pour protéger l'homme et les animaux.
Les informations fournies doivent découler et être corroborées par des données fournies pour la (les) substance(s) active(s) et celles visées aux sections 7 et 8.
4.3.1. Le cas échéant, spécifier les périodes d'attente avant la récolte, les périodes de réintroduction et les périodes de retrait nécessaires pour réduire au maximum la présence de résidus dans ou sur les récoltes, végétaux ou produits végétaux ou dans des espaces ou emplacements traités, en vue de protéger l'homme et les animaux, par exemple :
- délai d'attente avant la récolte (en jours) pour chaque culture concernée ;
- délai de réintroduction (en jours) du bétail dans les zones de pâturage ;
- délai de retour (en heures ou en jours) de l'homme dans les cultures, les bâtiments ou les espaces traités ;
- délai d'attente (en jours) avant usage pour l'alimentation des animaux ;
- période d'attente (en jours) entre l'application et la manipulation des produits traités ;
- délai d'attente (en jours) entre la dernière application et l'ensemencement ou la plantation des cultures suivantes.
4.3.2. Si nécessaire, compte tenu des résultats des essais, fournir des informations sur toute condition agricole, phytosanitaire ou environnementale particulière dans laquelle la préparation peut ou ne peut pas être utilisée.
4.4. Méthodes et précautions recommandées en matière de manipulation, d'entreposage, de transport ou en cas d'incendie
Indiquer les méthodes et les précautions recommandées concernant les techniques de manipulation (détaillées) pour le stockage, dans le magasin et chez l'utilisateur, des produits phytopharmaceutiques en vue de leur transport et en cas d'incendie. Fournir, lorsqu'elles existent, les informations relatives aux produits de combustion.
Spécifier les risques probables ainsi que les méthodes et procédures à appliquer en vue de minimiser les dangers. Indiquer les procédures à observer en vue de prévenir ou de minimiser la formation de déchets ou la rémanence du produit.
Le cas échéant, procéder à une évaluation selon ISO TR 9122.
Le cas échéant, indiquer la nature et les caractéristiques des vêtements et de l'équipement de protection proposés. Les informations fournies doivent permettre d'évaluer l'adéquation et l'efficacité de ceux-ci dans des conditions d'utilisation réalistes (par exemple dans les champs ou sous serres).
4.5. Mesures d'urgence en cas d'accident.
Si l'accident survient au cours du transport, du stockage ou de l'utilisation, indiquer les méthodes détaillées à suivre d'urgence ; ces méthodes comprennent :
- la contention des fuites ;
- la décontamination des terrains, véhicules et bâtiments ;
- l'élimination des emballages endommagés, des absorbants et autres matériaux ;
- la protection du personnel d'intervention et des assistants ;
- mesures de premiers secours en cas d'accident.
4.6. Procédures de destruction ou de décontamination du produit phytopharmaceutique et de son emballage.
Des procédures de destruction et de décontamination doivent être mises au point pour de petites quantités (niveau de l'utilisateur) et de grandes quantités (niveau du magasin). Les procédures doivent être conformes aux dispositions en vigueur concernant l'élimination des déchets, notamment toxiques. Les moyens d'élimination proposés ne doivent pas avoir d'incidence inacceptable sur l'environnement et représenter les moyens d'élimination les plus pratiques et les plus efficaces possible sur le plan des coûts.
4.6.1. Possibilité de les neutraliser.
Décrire les procédures de neutralisation (par exemple par réaction avec un alcali pour former des composés moins toxiques) à utiliser en cas de fuites accidentelles, si ces procédures sont envisageables. Evaluer, d'un point de vue pratique ou théorique, les produits obtenus après neutralisation et les préciser.
4.6.2. Incinération contrôlée.
Dans de nombreux cas, l'incinération contrôlée dans un incinérateur autorisé est le seul moyen d'éliminer en toute sécurité des substances actives ainsi que des produits phytopharmaceutiques contenant ces substances, des matériaux contaminés ou des emballages contaminés.
Si la teneur en halogènes de la (des) substances(s) actives(s) contenus dans la préparation est supérieure à 60 p. 100, indiquer le comportement pyrolitique de la substance active dans des conditions contrôlées (y compris, le cas échéant, l'apport d'oxygène et le temps de séjour fixé) à 800 °C et la teneur en dibenzo-p-dioxines polyhalogenatées et en dibenzo-furanes des produits de pyrolyse.
Le demandeur est tenu de fournir des instructions détaillées relatives à la sécurité de l'élimination.
4.6.3. Autres.
Décrire de manière exhaustive les autres méthodes d'élimination du produit phytopharmaceutique, emballages et matériaux contaminés, si elles sont proposées. Fournir des informations concernant ces méthodes en vue d'établir leur efficacité et sécurité.
5. Méthodes d'analyse.
(Arrêté du 1er février 1996, article 2 et Arrêté du 12 janvier 1998, article 2 I)
Introduction.
Les dispositions du présent point s'appliquent exclusivement aux méthodes d'analyse requises pour le contrôle et le suivi postérieurs à l'autorisation.
Pour les méthodes d'analyse utilisées pour la production des données requises par la présente directive ou à d'autres fins, le demandeur est tenu de fournir une justification de la méthode utilisée ; si nécessaire, des directives spécifiques seront mises au point pour de telles méthodes sur la base des mêmes normes que celles requises pour les méthodes de contrôle et de suivi postérieurs à l'autorisation.
La description des méthodes d'analyse doit être fournie et contenir toutes les données utiles concernant l'équipement, les matériels et les conditions d'application.
Ces méthodes doivent, autant que possible, suivre l'approche la plus simple, être peu onéreuses et faire appel à des équipements courants.
Les définitions suivantes s'appliquent aux fins du présent chapitre :
Impuretés : tout composant autre que la substance pure, dans la substance active technique (y compris les isomères non actifs) provenant du processus de fabrication ou de la dégradation survenue durant le stockage ;
Impuretés caractéristiques : impuretés posant des problèmes d'ordre toxicologique et/ou écotoxicologique ou environnemental ;
Métabolites : métabolites, y compris les produits résultant de la dégradation ou de la réaction de la substance active ;
Métabolites caractéristiques : métabolites posant des problèmes d'ordre toxicologique et/ou écotoxicologique ou environnemental.
A la demande, les échantillons suivants doivent être fournis :
i) des échantillons de la préparation ;
ii) un étalon pour l'analyse de la substance active pure ;
iii) un étalon de la substance active technique ;
iv) un étalon pour l'analyse des métabolites caractéristiques et/ou des autres composants compris dans la définition du résidu ;
v) si disponibles, des échantillons des substances de référence des impuretés importantes.
Définitions : voir annexe I, points 4.1 et 4.2.
5.1. Méthodes d'analyse de la préparation.
5.1.1. Il y a lieu de présenter et de décrire dans leur intégralité des méthodes qui permettent de doser la substance active présente dans la préparation. Lorsqu'une préparation contient plus d'une substance active, il y a lieu de présenter une méthode capable de doser chacune d'elles en présence des autres. Lorsqu'aucune méthode combinée n'est proposée, il y a lieu d'en fournir la justification technique. L'applicabilité des méthodes actuelles de la CIMAP doit être signalée.
5.1.2. Il convient également de présenter des méthodes qui permettent de doser dans la préparation les impuretés caractéristiques, si la composition de la préparation est telle que, sur la base d'une considération théorique, ces impuretés peuvent provenir du processus de fabrication ou de la dégradation survenue durant le stockage.
Le cas échéant, les méthodes de détermination des formulants de la préparation ou de leurs constituants doivent aussi être présentées.
5.1.3. Spécificité, linéarité, exactitude et répétabilité :
5.1.3.1. La spécificité des méthodes présentées doit être démontrée et mentionnée. Il y a lieu, en outre, de déterminer l'ampleur de l'interférence des autres substances présentes dans la préparation.
Les interférences d'autres composantes peuvent être considérées comme des erreurs systématiques dans l'évaluation de l'exactitude des méthodes proposées ; néanmoins une explication doit être donnée pour toute interférence contribuant pour plus de 3 % de la quantité totale dosée.
5.1.3.2. La linéarité des méthodes proposées dans une plage appropriée doit être déterminée et mentionnée. La plage d'étalonnage doit dépasser (d'au moins 20 %) la teneur nominale la plus élevée et la plus basse de la substance recherchée dans les solutions de la préparation. Pour l'étalonnage, on doit effectuer une double mesure d'au moins trois concentrations différentes ou une mesure simple de cinq concentrations. Les procès-verbaux doivent contenir l'équation de la courbe d'étalonnage et le coefficient de corrélation ainsi que des documents, relatifs à l'analyse, représentatifs et dûment étiquetés, par exemple des chromatogrammes.
5.1.3.3. Le critère d'exactitude n'est normalement applicable qu'aux méthodes de dosage de la substance active pure et des impuretés caractéristiques présentes dans la préparation.
5.1.3.4. Au moins cinq dosages sont normalement requis pour la répétabilité. L'écart type relatif (% ETR) doit être mentionné. Les valeurs aberrantes observées par une méthode appropriée (le test de Dixons ou de Grubbs, par exemple) peuvent être négligées, mais leur écart doit toujours être signalé et leur apparition doit faire l'objet d'une tentative d'explication.
5.2. Méthodes d'analyse pour le dosage des résidus.
Il y a lieu de présenter des méthodes d'analyse pour le dosage des résidus, sauf s'il est établi que les méthodes déjà proposées selon les exigences de l'annexe I, point 4.2, sont applicables.
Les mêmes dispositions que celles visées à l'annexe I, point 4.2, sont applicables.
6. Données relatives à l'efficacité I
(Arrêté du 1er février 1996, article 2)
Généralités.
Les données fournies doivent être suffisantes pour permettre une évaluation du produit phytopharmaceutique. Il doit être possible, en particulier, d'évaluer la nature et l'ampleur des avantages que procure l'utilisation de la préparation, par comparaison à des produits de référence et à des seuils d'infestation appropriés s'il en existe, et de définir ses conditions d'emploi.
Le nombre d'essais à effectuer et à relater dépend principalement de facteurs tels que l'étendue des connaissances relatives aux propriétés de la ou des substances actives que le produit contient ainsi que du nombre de situations rencontrées, y compris la variabilité des conditions phytosanitaires, les écarts climatiques, les diverses pratiques agricoles, l'uniformité des cultures, le mode d'application, le type d'organisme nuisible et le type de produit phytopharmaceutique.
Un nombre suffisant de données doit être produit et présenté en vue de confirmer que les modèles établis sont applicables dans les régions et pour la gamme de situations susceptibles de se présenter dans lesdites régions, pour lesquelles l'utilisation du produit doit être recommandée. Si un demandeur affirme que des essais dans une ou plusieurs des régions d'utilisation proposées sont superflus parce que la situation y est comparable avec celle d'autres régions où des essais ont été effectués, il doit étayer son affirmation de comparabilité à l'aide de preuves documentaires.
Pour évaluer d'éventuelles variations saisonnières, des données suffisantes doivent être produites et présentées en vue de confirmer l'efficacité des produits phytopharmaceutiques dans chaque région agronomique et climatique et pour chaque combinaison déterminée culture (ou production)/organismes nuisibles. Normalement, un compte rendu doit être effectué pour au moins deux campagnes d'essais relatifs à l'efficacité ou, s'il y a lieu, à la phytotoxicité.
Si, de l'avis du demandeur, les essais de la première campagne confirment bien la valeur des affirmations faites sur la base d'une extrapolation des résultats obtenus avec d'autres cultures, produits ou dans d'autres situations ou encore à partir d'autres essais effectués avec des préparations très voisines, il y a lieu de produire une justification, acceptable pour l'autorité compétente, de l'inutilité d'une seconde campagne. A l'inverse, si, en raison des conditions climatiques ou phytosanitaires ou pour d'autres raisons, les données obtenues dans une campagne déterminée sont d'une valeur limitée pour l'évaluation de l'efficacité, des essais au cours d'une ou plusieurs autres campagnes doivent être réalisés et relatés.
6.1. Essais préliminaires.
Des rapports, sous forme sommaire, concernant des essais préliminaires, y compris des études d'utilisation en serre ou en plein champ pour apprécier l'activité biologique et déterminer le dosage du produit phytopharmaceutique et de la ou des substances actives qu'il contient, doivent être présentés lorsque l'autorité compétente en fait la demande. Ces rapports donnent une information complémentaire à l'autorité compétente lorsqu'elle évalue le produit phytopharmaceutique. Si cette information n'est pas produite, il y a lieu de présenter une justification acceptable pour l'autorité compétente.
6.2. Essais d'efficacité.
But des essais.
Les essais doivent fournir des données suffisantes pour permettre une évaluation du niveau, de la durée et de l'uniformité du contrôle ou de la protection ou des autres effets attendus du produit phytopharmaceutique par comparaison avec des produits de référence appropriés s'il en existe.
Conditions d'essai.
Un essai comprend normalement trois paramètres : le produit à tester, le produit de référence et un témoin non traité.
L'action du produit phytopharmaceutique doit être examinée par rapport à des produits de référence appropriés s'il en existe. Un produit de référence approprié se définit comme un produit phytopharmaceutique autorisé qui s'est révélé suffisamment efficace dans la pratique et dans des conditions agronomiques, phytosanitaires et environnementales (y compris climatiques) existantes dans la région d'utilisation prévue. En général, le type de formation, les effets sur les organismes nuisibles, le spectre d'action et le mode d'application devraient être voisins de ceux du produit phytopharmaceutique testé.
Les produits phytopharmaceutiques doivent être testés dans des conditions où il a été démontré que l'organisme nuisible cible a été présent à un niveau qui produit ou est réputé produire des effets néfastes (rendement, qualité, résultat d'exploitation) sur une culture ou une superficie non protégée ou sur des végétaux ou produits végétaux qui n'ont pas été traités, ou que l'organisme nuisible est présent à un niveau tel qu'une évaluation du produit phytopharmaceutique peut être effectuée.
Les essais visant à fournir des données sur des produits phytopharmaceutiques destinés à la lutte contre les organismes nuisibles doivent démontrer leur degré d'efficacité sur les espèces d'organismes nuisibles en cause ou sur des espèces représentatives des groupes d'organismes nuisibles pour lesquels la demande est présentée. Les essais doivent porter sur les différents stades de croissance ou, s'il y a lieu, sur le cycle de vie des espèces nuisibles, ainsi que sur les diverses souches ou races si celles-ci sont susceptibles de présenter des degrés de sensibilité différents.
De même, les essais visant à fournir des données sur les produits phytopharmaceutiques qui sont des régulateurs de croissance doivent démontrer leur degré d'efficacité sur l'espèce à traiter et inclure une étude sur les différentes réactions d'un échantillon représentatif de la gamme des variétés cultivées pour le traitement desquelles le produit est proposé.
En vue de déterminer la réaction aux différentes doses, il est nécessaire de procéder à des essais à des doses inférieures à la dose recommandée en vue de déterminer si la dose recommandée est la dose minimale nécessaire pour obtenir l'effet voulu.
La durée des effets du traitement doit être étudiée en rapport avec la lutte contre l'organisme cible ou l'effet sur les végétaux ou produits végétaux traités, selon le cas. Lorsque plus d'une application est recommandée, il y a lieu de relater les essais établissant la durée des effets d'une application, le nombre d'applications nécessaires et les intervalles souhaités entre applications.
Des preuves doivent être fournies en vue de démontrer que la dose, l'époque et le mode d'application recommandés donnent des résultats adéquats en matière de lutte ou de protection ou qu'ils produisent l'effet voulu dans toutes les situations et utilisations pratiques probables.
Sauf si des indices précis donnent à penser que l'action du produit phytopharmaceutique ne sera probablement pas réduite de manière significative par des facteurs liés à l'environnement, tels que la température ou les précipitations, une enquête sur les effets de tels facteurs sur l'action du produit doit être effectuée et relatée, en particulier s'il est notoire que l'action de produits chimiques voisins s'en trouve réduite.
Lorsque les mentions figurant sur l'étiquette comprennent des recommandations relatives à l'emploi du produit phytopharmaceutique avec un ou plusieurs autres produits phytopharmaceutiques ou adjuvants, des informations doivent être données quant au résultat escompté du mélange.
Ligne directrice pour les essais.
Les essais doivent être conçus en vue d'étudier certains points particuliers, de limiter autant que possible les effets d'une variation aléatoire entre les différentes parties d'un même site et de permettre une analyse statistique des résultats. La conception, l'analyse et le rapport des essais doivent être conformes aux lignes directrices 152 et 181 de l'Organisation européenne et méditerranéenne pour la protection des plantes (O.E.P.P.). Le rapport doit comporter une évaluation critique et détaillée des données.
Les essais doivent être effectués conformément aux lignes directrices O.E.P.P. spécifiques, si elles existent, ou, lorsqu'un Etat membre l'exige et que l'essai est réalisé sur le territoire de cet Etat membre, conformément à des lignes directrices répondant au moins aux exigences de la ligne directrice O.E.P.P. correspondante.
Les résultats dont l'analyse statistique est pertinente doivent faire l'objet d'une telle analyse ; cela peut nécessiter une adaptation de la ligne directrice.
6.3. Informations sur l'apparition ou le développement éventuel d'une résistance.
Les données de laboratoire et, si elles existent, les informations recueillies sur le terrain en ce qui concerne l'apparition et le développement d'une résistance ou d'une résistance croisée, dans des populations d'organismes nuisibles, à la ou aux substances actives ou à des substances actives connexes doivent être fournies. Même si ces informations ne concernent pas directement les utilisations pour lesquelles l'autorisation est demandée ou doit être renouvelée (différentes espèces d'organismes nuisibles ou différentes cultures), elles doivent être fournies si elles sont disponibles parce qu'elles peuvent donner une indication de la probabilité du développement d'une résistance dans la population cible.
S'il existe des éléments de preuve ou des informations suggérant que, dans des conditions d'utilisation commerciale, le développement d'une résistance est probable, des preuves doivent être recueillies et présentées en ce qui concerne la sensibilité de la population de l'organisme nuisible en cause au produit phytopharmaceutique. Il y a lieu de fournir en pareil cas une stratégie de gestion destinée à réduire au strict minimum le développement d'une résistance ou d'une résistance croisée dans la population cible.
6.4. Incidences du traitement sur le rendement et/ou la qualité des végétaux ou produits végétaux.
6.4.1. Incidences sur la qualité des végétaux ou produits végétaux.
But des essais.
Les essais doivent fournir des données suffisantes pour permettre une évaluation de l'apparition possible d'un changement du goût ou de l'odeur, ou d'autres aspects qualitatifs de végétaux ou produits végétaux, après traitement à l'aide du produit phytopharmaceutique.
Situations dans lesquelles les essais sont requis.
La possibilité d'apparition d'un changement du goût ou de l'odeur dans les produits végétaux alimentaires doit être recherchée et relatée :
- lorsque la nature du produit ou son utilisation est telle qu'un risque d'un changement du goût ou de l'odeur est à prévoir,
ou
- lorsque d'autres produits à base de la même substance active ou d'une substance très similaire se sont révélés susceptibles de produire un changement du goût ou de l'odeur.
Les effets des produits phytopharmaceutiques sur d'autres aspects qualitatifs des végétaux ou produits végétaux traités doivent être déterminés et relatés :
- lorsque la nature du produit phytopharmaceutique ou son utilisation pourrait avoir une incidence néfaste sur d'autres aspects qualitatifs (par exemple en cas d'utilisation de régulateurs de croissance peu avant la récolte),
ou
- lorsque d'autres produits fabriqués à partir de la même substance active ou d'une substance très similaire se sont révélés avoir une influence néfaste sur la qualité.
Il convient de réaliser les essais en premier lieu sur les principales cultures auxquelles le produit phytopharmaceutique est destiné, en doublant la dose normale d'utilisation et en recourant si possible aux méthodes de traitement les plus courantes. Si des effets sont observés, il est nécessaire d'effectuer les essais à la dose normale d'application.
L'étendue des recherches nécessaires sur d'autres cultures dépendra de leur degré de similitude avec les cultures principales déjà testées, de la quantité et de la qualité des données disponibles sur ces cultures principales ainsi que du degré de similitude entre les modes d'utilisation du produit phytopharmaceutique et, s'il y a lieu, entre les méthodes de traitement des cultures. Il suffit généralement d'effectuer l'essai avec la principale formulation à autoriser.
6.4.2. Incidences sur les processus de transformation.
But des essais.
Les essais doivent fournir des données suffisantes pour permettre une évaluation de l'apparition éventuelle d'effets néfastes, après traitement au moyen du produit phytopharmaceutique, sur les processus de transformation ou sur la qualité des produits issus de la transformation.
Situations dans lesquelles les essais sont requis.
Lorsque les végétaux ou produits végétaux traités sont normalement destinés à être utilisés dans un processus de transformation tel que la vinification, la fabrication de la bière ou la panification, et en présence de résidus de récolte significatifs, l'éventualité de l'apparition d'effets néfastes doit être examinée et relatée :
- lorsque certains indices tendent à prouver que l'utilisation du produit phytopharmaceutique pourrait avoir une influence sur les procédés appliqués (par exemple en cas d'utilisation de régulateurs de croissance ou de fongicides peu de temps avant la récolte),
ou
- lorsque d'autres produits fabriqués à partir de la même substance active ou d'une substance très similaire se sont révélés avoir une influence néfaste sur ces processus ou sur les produits issus de la transformation.
Il suffit généralement d'effectuer l'essai avec la principale formulation à autoriser.
6.4.3. Effets sur le rendement des végétaux ou produits végétaux traités.
But des essais.
Les essais doivent fournir des données suffisantes pour permettre une évaluation de l'action du produit phytopharmaceutique et de l'apparition possible d'une baisse de rendement ou d'une perte au stockage des végétaux ou produits végétaux traités.
Situations dans lesquelles les essais sont requis.
L'incidence des produits phytopharmaceutiques sur le rendement ou les composantes du rendement des végétaux ou produits végétaux traités doit être déterminée le cas échéant. Si les végétaux ou produits végétaux traités sont appelés à être stockés, l'effet sur le rendement après stockage, y compris les données sur la durée de stockage, doit être déterminé le cas échéant.
Cette information est normalement fournie par les essais requis en vertu des dispositions du point 6.2.
6.5. Phytotoxicité pour les végétaux cibles (y compris différents cultivars) ou les produits végétaux cibles.
But des essais.
Les essais doivent fournir des données suffisantes pour permettre une évaluation de l'action du produit phytopharmaceutique et d'une éventuelle phytotoxicité après traitement à l'aide du produit phytopharmaceutique.
Situations dans lesquelles les essais sont requis.
Pour les herbicides et autres produits phytopharmaceutiques donnant lieu à l'apparition d'effets néfastes, quoique temporaires, pendant les essais effectués conformément au point 6.2, les marges de sélectivité sur des cultures cibles doivent être établies par l'application d'une dose double de la dose recommandée. Si de graves effets de phytotoxicité apparaissent, l'essai doit aussi être effectué avec une dose intermédiaire.
Si des effets néfastes se produisent, mais qu'ils sont considérés comme négligeables au regard des avantages que procure l'utilisation du produit ou comme passagers, la validité de cette affirmation doit être démontrée. Il peut y avoir lieu de produire des mesures de rendement.
L'innocuité d'un produit phytopharmaceutique à l'égard des principaux cultivars des principales cultures pour lesquelles il est recommandé doit être démontrée ; cela concerne notamment les effets du stade de croissance, la vigueur ainsi que d'autres facteurs susceptibles d'influencer la sensibilité à l'endommagement.
L'étendue des recherches nécessaires sur d'autres cultures dépendra de leur degré de similitude avec les cultures principales déjà testées, de la quantité et de la qualité des données disponibles sur ces cultures principales et, s'il y a lieu, du degré de similitude entre les modes d'utilisation du produit phytopharmaceutique. Il suffit généralement d'effectuer l'essai avec la principale formulation à autoriser.
Si les indications figurant sur l'étiquette comportent des recommandations relatives à l'utilisation du produit phytopharmaceutique avec un ou plusieurs produits phytopharmaceutiques ou adjuvants, les dispositions des paragraphes ci-dessus s'appliquent au mélange.
Ligne directrice pour l'essai.
Les observations concernant la phytotoxicité doivent être faites dans les essais prévus au point 6.2.
Si des effets de phytotoxicité sont observés, ils doivent être déterminés avec précision et faire l'objet d'un rapport conformément à la ligne directrice O.E.P.P. 135 ou, lorsqu'un Etat membre l'exige et que l'essai est réalisé sur le territoire de cet Etat, conformément à des lignes directrices répondant au moins aux exigences de cette ligne directrice.
Les résultats dont l'analyse statistique est pertinente doivent faire l'objet d'une telle analyse ; cela peut nécessiter une adaptation de la ligne directrice.
6.6. Observations concernant les effets secondaires indésirables ou non recherchés, par exemple sur des organismes utiles ou d'autres organismes non ciblés, sur les cultures suivantes, sur d'autres végétaux ou parties de végétaux traités et utilisés à des fins de multiplication (par exemple semences, boutures, stolons).
6.6.1. Incidence sur les cultures suivantes.
But de l'information requise.
Des données suffisantes doivent être fournies pour permettre une évaluation des éventuels effets néfastes d'un traitement à l'aide d'un produit phytopharmaceutique sur les cultures successives.
Situations dans lesquelles l'information est requise.
Si les données obtenues conformément à la section 9, point 9.1, indiquent que des quantités significatives de résidus de la substance active, de ses métabolites ou produits de dégradation, ayant ou pouvant avoir une activité biologique sur les cultures suivantes, subsistent dans le sol ou dans les substances végétales telles que la paille ou la matière organique jusqu'au stade du semis ou de la plantation d'éventuelles cultures suivantes, des observations doivent être faites quant aux effets de ces produits sur la gamme normale des cultures suivantes.
6.6.2. Incidence sur d'autres végétaux, y compris les cultures limitrophes.
But de l'information requise.
Des données suffisantes doivent être fournies pour permettre une évaluation des éventuels effets néfastes d'un traitement à l'aide du produit phytopharmaceutique sur d'autres végétaux, et notamment sur les cultures limitrophes.
Situations dans lesquelles l'information est requise.
Des observations doivent être présentées en ce qui concerne les effets néfastes sur d'autres végétaux, et notamment sur la gamme normale des cultures limitrophes, lorsqu'il y a lieu de considérer que le produit phytopharmaceutique peut toucher ces végétaux par déplacement de vapeurs.
6.6.3. Incidence sur les végétaux ou produits végétaux traités à utiliser à des fins de multiplication.
But de l'information requise.
Des données suffisantes doivent être fournies pour permettre une évaluation des éventuels effets néfastes d'un traitement à l'aide du produit phytopharmaceutique sur les végétaux ou produits végétaux à utiliser à des fins de multiplication.
Situations dans lesquelles l'information est requise.
Des observations doivent être présentées en ce qui concerne l'incidence des produits phytopharmaceutiques sur les parties de végétaux utilisées à des fins de multiplication, sauf si les utilisations proposées excluent les cultures destinées à la production de semences, de boutures, de stolons ou de tubercules destinés à la plantation :
i) Semences : viabilité, germination et vigueur ;
ii) Boutures : enracinement et taux de reprise ;
iii) Stolons : implantation et taux de reprise ;
iv) Tubercules : germination et croissance normale.
Ligne directrice pour l'essai.
Les essais de semences s'effectuent selon les méthodes A.I.E.S. (3).
6.6.4. Tout effet, positif ou négatif, sur l'incidence d'autres organismes nuisibles, observé dans le cadre d'essais effectués conformément aux conditions de la présente section doit être relaté. Toute incidence observée sur l'environnement, et notamment l'incidence sur la faune et/ou les organismes utiles, doit également être relatée.
6.7. Résumé et évaluation des données fournies au titre des points 6.1 à 6.6.
Un résumé de toutes les données et informations fournies au titre des points 6.1 à 6.6 doit être fourni conjointement avec une évaluation détaillée et critique des données, axée sur les avantages offerts par le produit phytopharmaceutique, sur ses effets néfastes avérés ou probables ainsi que sur les mesures nécessaires pour les prévenir ou les réduire au strict minimum.
7. Etudes de toxicité
(Arrêté du 1er février 1996, article 2 et Arrêté du 12 janvier 1998, article 2 II)
Pour une bonne évaluation de la toxicité des préparations, des informations suffisantes sur la toxicité aigue, l'irritation et la sensibilisation causées par la substance active doivent être disponibles. Si possible, des informations supplémentaires sur le mode d'action toxique, le profil toxicologique et tout autre aspect toxicologique connu de la substance active doivent être présentées.
Compte tenu de l'influence que les impuretés et d'autres composants peuvent exercer sur le comportement toxicologique, il est essentiel que, pour toute étude proposée, une description détaillée (spécification) du matériel utilisé soit fournie. Des tests doivent être effectués avec le produit phytopharmaceutique à autoriser.
7.1. Toxicité aigue.
Les études, données et informations à fournir et à évaluer doivent être suffisantes pour permettre de déceler les effets d'une exposition unique au produit phytopharmaceutique, et en particulier pour déterminer ou indiquer :
- la toxicité du produit phytopharmaceutique ;
- la toxicité du produit phytopharmaceutique par rapport à la substance active ;
- l'évolution au cours du temps et les caractéristiques de l'effet avec des détails complets sur les modifications comportementales et les résultats macropathologiques éventuels d'un examen post mortem ;
- si possible, le mécanisme d'action toxique ;
- le danger relatif lié aux diverses voies d'exposition.
Si l'accent doit être placé sur l'estimation des niveaux de toxicité considérés, les informations obtenues doivent aussi permettre de classer le produit phytopharmaceutique selon la directive 78/631/CEE. Les informations obtenues grâce aux essais de toxicité aiguë ont une valeur particulière pour l'évaluation des dangers que risquent de présenter des cas accidentels.
7.1.1. Orale.
Situations dans lesquelles l'essai est requis :
Un essai de toxicité aiguë par voie orale doit toujours être effectué, sauf si le demandeur peut justifier, à la satisfaction de l'autorité compétente, que l'article 3, paragraphe 2, de la directive 78/631/CEE peut être invoqué.
Ligne directrice pour l'essai :
L'essai doit être effectué conformément à la directive 92/69/CEE, méthode B 1 ou B 1 bis.
7.1.2. Dermale.
Situations dans lesquelles l'essai est requis :
Un essai de toxicité aiguë par voie dermale doit toujours être effectué, sauf si le demandeur peut justifier, à la satisfaction de l'autorité compétente, que l'article 3, paragraphe 2, de la directive 78/631/CEE peut être invoqué.
Ligne directrice pour l'essai :
L'essai doit être effectué conformément à la directive 92/69/CEE, méthode B 3.
7.1.3. Inhalatoire.
But de l'essai :
L'essai doit déterminer la toxicité par inhalation, pour les rats, d'un produit phytopharmaceutique ou de la fumée qu'il dégage.
Situations dans lesquelles l'essai est requis :
L'essai doit être effectué lorsque le produit phytopharmaceutique :
- est un gaz, notamment liquéfié ;
- est une préparation fumigène ou un fumigant ;
- est utilisé à l'aide d'un équipement de nébulisation ;
- est une préparation produisant de la vapeur ;
- est un aérosol ;
- est une poudre contenant une proportion significative de particules d'un diamètre 50 micro m ( 1 p. 100 sur la base du poids) ;
- est appliqué par aéronef dans le cas où l'exposition par inhalation est pertinente ;
- lorsque le produit phytopharmaceutique contient une substance active dont la pression de vapeur 1 x 10-2 Pa et doit être utilisé dans des espaces clos tels que des magasins ou des serres ;
- doit être appliqué selon un procédé produisant une proportion significative de particules ou gouttelettes d'un diamètre 50 micro m (1 p. 100 sur la base du poids).
Ligne directrice pour l'essai ;
L'essai doit être effectué conformément à la directive 92/69/CEE, méthode B 2.
7.1.4. Irritation de la peau.
But de l'essai :
L'essai doit permettre de mettre en évidence le pouvoir irritant pour la peau du produit phytosanitaire, y compris la réversibilité potentielle des effets observés.
Situations dans lesquelles l'essai est requis :
Le pouvoir d'irritation pour la peau du produit phytopharmaceutique doit être déterminé sauf si, comme il est indiqué dans la ligne directrice pour l'essai, il est probable que des effets graves sur la peau peuvent se produire ou que ces effets peuvent être exclus.
Ligne directrice pour l'essai :
L'essai doit être effectué conformément à la directive 92/69/CEE, méthode B 4.
7.1.5. Irritation des yeux.
But de l'essai :
L'essai doit permettre de mettre en évidence le pouvoir irritant pour les yeux du produit phytopharmaceutique, y compris la réversibilité potentielle des effets observés.
Situations dans lesquelles l'essai est requis :
Les essais d'irritation des yeux doivent être effectués sauf s'il est probable, conformément à la ligne directrice pour l'essai, que les essais peuvent être gravement nocifs pour les yeux.
Ligne directrice pour l'essai :
L'irritation des yeux doit être déterminée conformément à la directive 92/69/CEE, méthode B 5.
7.1.6. Sensibilisation de la peau.
But de l'essai :
L'essai fournira des informations suffisantes pour évaluer le pouvoir du produit phytopharmaceutique de provoquer des réactions de sensibilisation de la peau.
Situations dans lesquelles les essais sont requis :
Les essais doivent toujours être effectués sauf si la (les) substance(s) active(s) ou les coformulants sont reconnus pour avoir un pouvoir de sensibilisation.
Ligne directrice pour les tests :
Les tests doivent être effectués conformément à la directive 92/69/CEE, méthode B 6.
7.1.7. Etudes complémentaires pour des combinaisons de produits phytopharmaceutiques.
But de l'essai :
Dans certains cas, il peut être nécessaire d'effectuer les études visées aux points 7.1.1 à 7.1.6 pour une combinaison de produits phytopharmaceutiques, lorsque l'étiquette du produit comporte des indications d'utilisation du produit phytopharmaceutique avec d'autres produits phytopharmaceutiques et/ou avec des adjuvants mélangés dans le réservoir de l'appareil de pulvérisation. Les décisions relatives à la nécessité d'études complémentaires doivent être prises cas par cas compte tenu des résultats des études de toxicité aiguë relatives aux différents produits phytopharmaceutiques, de la possibilité d'exposition à la combinaison de produits en cause et des informations disponibles ou de l'expérience pratique concernant les produits en cause ou des produits similaires.
7.2. Données relatives à l'exposition.
Pour la mesure de l'exposition à un produit phytopharmaceutique dans l'air que respirent les opérateurs, les personnes présentes ou les travailleurs, les exigences relatives aux méthodes de mesure décrites à l'annexe II bis de la directive 80/1107/CEE du Conseil du 27 novembre 1980, concernant la protection des travailleurs contre les risques liés à une exposition pendant le travail, doivent être prises en considération.
7.2.1. Exposition de l'opérateur.
Les risques des produits phytopharmaceutiques pour les utilisateurs dépendent des propriétés physiques, chimiques et toxicologiques du produit phytopharmaceutique ainsi que du type de produit (non dilué/dilué) et de la voie, du degré et de la durée d'exposition. Des informations et des données suffisantes doivent être produites et relatées pour permettre une évaluation de l'intensité d'exposition à la (aux) substance(s) active(s) et/ou aux composés toxicologiquement importants du produit phytopharmaceutique susceptible de se produire dans les conditions d'utilisation proposées. Elles doivent aussi fournir une base de sélection des mesures de protection appropriées, y compris l'équipement de protection individuelle à utiliser par les opérateurs et à indiquer sur l'étiquette.
7.2.1.1. Estimation de l'exposition de l'opérateur.
But de l'estimation :
Une estimation doit être faite grâce à l'utilisation d'un modèle de calcul approprié, s'il existe, devant permettre une évaluation de l'exposition à laquelle l'opérateur sera probablement soumis dans les conditions d'utilisation proposées.
Situations dans lesquelles l'estimation est requise :
Une estimation de l'exposition de l'opérateur doit toujours être effectuée.
Conditions d'estimation :
Une estimation doit être faite pour toute méthode d'application et tout type d'équipement d'application proposés pour le produit phytopharmaceutique compte tenu des exigences résultant de l'application des dispositions prévues par la directive 78/631/CEE en matière de classement et d'étiquetage pour la manipulation du produit éventuellement dilué ainsi que des différents types et tailles de récipients à utiliser, des opérations de mélange et de chargement, de l'application du produit phytopharmaceutique, des conditions climatiques et du nettoyage et de l'entretien habituel de l'équipement d'application.
Tout d'abord, une estimation doit être faite dans l'hypothèse où l'opérateur n'utilise aucun équipement de protection individuelle.
Le cas échéant, une seconde estimation doit être faite dans l'hypothèse où l'opérateur utilise un équipement de protection efficace et disponible sur le marché. Si les mesures de protection sont spécifiées sur l'étiquette, l'estimation en tiendra compte.
7.2.1.2. Mesure de l'exposition de l'opérateur.
But de l'essai :
L'essai doit fournir des données suffisantes pour permettre une évaluation de l'exposition que l'opérateur est susceptible de subir dans les conditions d'utilisation proposées.
Situations dans lesquelles l'essai est requis :
Les données effectives d'exposition concernant la (les) principale(s) voie(s) d'exposition doivent être relatées si l'évaluation du risque indique qu'une valeur limite concernant la santé est dépassée. Cela est, par exemple, le cas si les résultats de l'estimation relative à l'exposition de l'opérateur fournis au point 7.2.1.1 indiquent que :
- le(s) niveau(x) acceptable(s) d'exposition de l'opérateur (NAEO) fixé(s) en cas d'inscription de la (des) substance(s) active(s) à l'annexe I peut (peuvent) être dépassé(s) ;
- les valeurs limites fixées pour la substance active et/ou le (les) composé(s) toxicologiquement important(s) des produits phytopharmaceutiques conformément à la directive 80/1107/CEE concernant la protection des travailleurs contre les risques liés à une exposition à des agents chimiques, physiques et biologiques pendant le travail, et conformément à la directive 90/394/CEE du Conseil du 28 juin 1990 concernant la protection des travailleurs contre les risques liés à l'exposition à des agents cancérigènes au travail, peuvent être dépassées.
Les données réelles d'exposition doivent aussi être relatées lorsque aucun modèle de calcul approprié ni aucune donnée appropriée ne sont disponibles pour effectuer l'estimation prévue au point 7.2.1.1.
Dans les cas où l'exposition cutanée est la voie d'exposition principale, un essai d'absorption par la peau ou une étude de toxicité dermale subaiguë, s'ils ne sont pas déjà disponibles, peuvent être des essais de remplacement utiles pour fournir les données nécessaires pour affiner l'estimation prévue au point 7.2.1.1.
Conditions d'essai :
L'essai doit être effectué dans des conditions d'exposition réalistes tenant compte des conditions d'utilisation proposées.
7.2.2. Exposition des personnes présentes.
Les personnes présentes peuvent être exposées pendant l'application des produits phytopharmaceutiques. Des informations et des données suffisantes doivent être relatées pour fournir une base de sélection des conditions d'utilisation appropriées, y compris l'interdiction des personnes présentes sur le lieu du traitement et les distances à respecter.
But de l'estimation :
Une estimation doit être faite à l'aide d'un modèle de calcul approprié, s'il existe, pour permettre une évaluation de l'exposition probable des personnes présentes dans les conditions d'utilisation proposées.
Situations dans lesquelles l'estimation est requise :
Une estimation de l'exposition des personnes présentes doit toujours être effectuée.
Conditions d'estimation :
Une estimation de l'exposition des personnes présentes doit être faite pour chaque méthode d'application. L'estimation doit être faite dans l'hypothèse où les personnes présentes ne portent aucun équipement de protection individuelle.
Des mesures d'exposition de la personne présente types peuvent être exigées lorsque les estimations font état d'une situation préoccupante.
7.2.3. Exposition des travailleurs.
Les travailleurs peuvent être exposés à la suite de l'application de produits phytopharmaceutiques, en pénétrant sur des terres ou dans des locaux traités ou en manipulant des végétaux ou des produits végétaux traités sur lesquels persistent des résidus. Des informations et des données suffisantes doivent être relatées pour fournir une base de sélection des dispositions de protection appropriées, y compris les périodes d'attente et d'exclusion des lieux.
7.2.3.1. Estimation de l'exposition des travailleurs.
But de l'estimation :
Une estimation doit être faite sur la base d'un modèle de calcul approprié, si ce modèle existe, afin de permettre une évaluation de l'exposition des travailleurs susceptible de se produire dans les conditions d'utilisation proposées.
Situations dans lesquelles l'estimation est requise :
Une estimation de l'exposition des travailleurs doit toujours être effectuée.
Conditions d'estimation :
Une estimation d'exposition des travailleurs doit être faite pour toute culture et pour toute tâche à effectuer.
Tout d'abord, l'estimation doit être faite sur la base des données disponibles concernant l'exposition escomptée dans l'hypothèse où le travailleur n'utilise pas d'équipement de protection individuelle.
Le cas échéant, une deuxième estimation doit être faite dans l'hypothèse où le travailleur utilise un équipement de protection efficace, disponible sur le marché.
Le cas échéant, une autre estimation doit être faite sur la base de données obtenues concernant la quantité de résidus désadsorbables dans les conditions d'utilisation proposées.
7.2.3.2. Mesure de l'exposition des travailleurs :
But de l'essai :
L'essai doit fournir des données suffisantes pour permettre une évaluation de l'exposition probable des travailleurs dans les conditions d'utilisation proposées.
Situations dans lesquelles l'essai est requis :
Les données relatives à l'exposition réelle par la (les) voie(s) d'exposition principale(s) doivent être relatées lorsque l'évaluation du risque indique qu'une valeur limite concernant la santé est dépassée. Cela est, par exemple, le cas lorsque les résultats de l'estimation de l'exposition des travailleurs visée au point 7.2.3.1 indiquent que :
- les valeurs limites fixées pour la substance active et/ou le (les) composé(s) toxicologiquement important(s) des produits phytopharmaceutiques conformément à la directive 80/1107/CEE concernant la protection des travailleurs contre les risques liés à une exposition à des agents chimiques, physiques et biologiques pendant le travail, et conformément à la directive 90/394/CEE concernant la protection des travailleurs contre les risques liés à l'exposition à des agents cancérigènes au travail peuvent être dépassées.
Les données réelles d'exposition doivent aussi être relatées lorsqu'aucun modèle de calcul approprié ni aucune donnée appropriée ne sont disponibles pour effectuer l'estimation prévue au point 7.2.3.1.
Dans les cas où l'exposition cutanée est la voie d'exposition principale, un essai d'absorption par la peau peut, s'il n'est pas déjà disponible, être un essai de remplacement utile pour fournir les données nécessaires pour affiner l'estimation prévue au point 7.2.3.1.
Conditions d'essai :
L'essai doit être effectué dans des conditions d'exposition réalistes tenant compte des conditions d'utilisation proposées.
7.3. Absorption cutanée.
But de l'essai :
L'essai doit donner une mesure de l'absorption par la peau de la substance active et des composés toxicologiquement importants.
Situations dans lesquelles l'essai est requis :
L'étude doit être effectuée lorsque l'exposition cutanée est une voie d'exposition significative et que l'évaluation du risque indique qu'une valeur limite concernant la santé est dépassée. Cela est, par exemple, le cas lorsque les résultats de l'estimation ou de la mesure de l'exposition de l'opérateur visées aux points 7.2.1.1 ou 7.2.1.2 indiquent que :
- le(s) niveau(x) acceptable(s) d'exposition de l'opération (NAEO) fixé(s) en cas d'inscription de la (des) substance(s) active(s) peu(peuvent) être dépassé(s) ;
- les valeurs limites fixées pour la substance active et/ou le(les) composé(s) toxicologiquement important(s) des produits phytopharmaceutiques conformément à la directive 80/1107/CEE concernant la protection des travailleurs contre les risques liés à une exposition à des agents chimiques, physiques et biologiques pendant le travail, et conformément à la directive 90/394/CEE concernant la protection des travailleurs contre les risques liés à l'exposition à des agents cancérigènes au travail, peuvent être dépassées.
Conditions d'essai :
En principe, les données d'une étude d'absorption cutanée in vivo sur le rat doivent être relatées. Si, lorsque les résultats de l'estimation utilisant les données relatives à l'absorption cutanée in vivo sont incorporés dans l'évaluation du risque, il subsiste une indication d'exposition excessive, il peut être nécessaire de réaliser une étude d'absorption comparative in vitro sur le rat et la peau humaine.
Ligne directrice pour l'essai :
Utiliser des éléments appropriés de la directive 417 de l'O.C.D.E. En ce qui concerne la conception des études, il peut être nécessaire de tenir compte des résultats des études d'absorption par la peau de la(des) substance(s) active(s).
7.4. Données toxicologiques disponibles concernant des substances non actives.
Si elle est disponible, présenter une copie de la notification et de la fiche de données relatives à la sécurité dans le cadre de la directive 67/548/CEE et de la directive 91/155/CEE de la Commission, du 5 mars 1991, définissant et fixant, en application de l'article 10 de la directive 88/379/CEE du Conseil, les modalités du système d'information spécifique relatif aux préparations dangereuses pour chacun des autres composants. Toute autre information disponible devrait être présentée.
8. Résidus dans ou sur les produits traités, les denrées alimentaires et les aliments pour animaux.
Introduction.
Les dispositions de l'introduction du point 6 de l'annexe I sont applicables.
8.1. Métabolisme, distribution et expression du résidu dans les végétaux et les animaux d'élevage.
But des essais.
Les objectifs des présentes études sont les suivants :
- donner une estimation des résidus finaux totaux se trouvant dans la fraction pertinente des produits de la récolte qui ont été traités selon le programme prévu ;
- quantifier le taux de dégradation et d'excrétion des résidus totaux dans certains produits animaux (lait ou œufs) et excrétions animales ;
- identifier les principaux composants des résidus finaux totaux présents dans les produits de la récolte et dans les produits animaux comestibles ;
- indiquer la distribution des résidus entre les fractions pertinentes des produits à la récolte et entre les produits animaux comestibles pertinents ;
- quantifier les principaux composants du résidu et démontrer l'efficacité des méthodes d'extraction de ces composants ;
- fournir des données sur lesquelles on peut fonder une décision relative à la nécessité d'effectuer des études sur l'alimentation du bétail conformément au point 8.3 ;
- décider de la définition et de l'expression d'un résidu.
Situations dans lesquelles les essais sont requis.
Des essais complémentaires sur le métabolisme ne doivent être effectués que s'il n'est pas possible de procéder à une extrapolation à partir des données obtenues au sujet de la substance active, conformément aux conditions de l'annexe I, points 6.1 et 6.2. Cela peut être le cas pour des produits végétaux ou des animaux d'élevage pour lesquels des données n'ont pas été proposées dans le cadre de l'insertion de la substance active à l'annexe I ou n'étaient pas requises pour modifier les conditions de son insertion dans la liste communautaire des substances actives ou dans le cas où l'on pourrait escompter un métabolisme différent.
Conditions des essais.
Les mêmes dispositions que celles prévues à l'annexe I, points 6.1 et 6.2, sont applicables.
8.2. Essais relatifs aux résidus.
But des essais.
Les objectifs des présentes études sont les suivants :
- quantifier les concentrations de résidus maximales probables dans les cultures traitées au moment de leur récolte ou de la sortie du stock conformément aux bonnes pratiques agricoles proposées, et
- déterminer, le cas échéant, le rythme de diminution des dépôts du produit phytopharmaceutique.
Situations dans lesquelles les essais sont requis.
Des essais complémentaires de détermination des résidus ne doivent être effectués que s'il n'est pas possible de procéder à une extrapolation à partir des données obtenues au sujet de la substance active conformément aux conditions de l'annexe I, point 6.3. Cela peut être le cas pour des formules spéciales, pour des méthodes spéciales d'application ou pour des produits végétaux pour lesquels des données n'avaient pas été présentées dans le cadre de l'insertion de la substance active dans la liste communautaire des substances actives ou n'étaient pas requises pour modifier les conditions de son insertion dans la liste communautaire des substances actives.
Conditions des essais.
Les mêmes dispositions que celles prévues à l'annexe I, point 6.3, sont applicables.
8.3. Etudes sur l'alimentation des animaux.
But des essais.
L'objectif de ces études est de déterminer le taux de résidus contenus dans les produits animaux et provenant des résidus contenus dans les aliments pour animaux ou cultures fourragères.
Situations dans lesquelles les essais sont requis.
Des études complémentaires relatives à l'alimentation des animaux en vue d'évaluer les limites maximales de résidus dans les produits animaux ne sont nécessaires que s'il n'est pas possible de procéder à une extrapolation à partir des données obtenues sur la substance active conformément aux dispositions de l'annexe I, point 6.4. Tel peut être le cas lorsque des cultures fourragères supplémentaires doivent être autorisées, entraînant une ingestion supplémentaire de résidus par le bétail pour lesquels des données n'avaient pas été présentées en vue de l'insertion de la substance dans la liste communautaire des substances actives ou n'étaient pas nécessaires pour modifier les conditions de son insertion dans la liste communautaire de substances actives.
Conditions des essais.
Les mêmes conditions que celles prévues à l'annexe I, point 6.4, sont applicables.
8.4. Effets de la transformation industrielle et/ou des préparations domestiques.
But des essais.
Les principaux objectifs des présentes études sont les suivants :
- déterminer si la présence de résidus dans les produits crus entraîne ou non la formation de produits de dégradation ou de réaction pendant la transformation, ce qui peut nécessiter une évolution séparée du risque ;
- déterminer la distribution quantitative des résidus dans les divers produits intermédiaires et finis et estimer les facteurs de transfert ;
- permettre une estimation plus réaliste de l'ingestion de résidus par la ration alimentaire ou fourragère.
Situations dans lesquelles les essais sont requis.
Des études supplémentaires ne doivent être effectuées que s'il n'est pas possible de procéder à une extrapolation à partir des données obtenues pour la substance active conformément aux dispositions de l'annexe I, point 6.5. Tel peut être le cas pour des produits végétaux pour lesquels des données n'avaient pas été présentées dans le cadre de l'insertion de la substance active dans la liste communautaire des substances actives ou n'étaient pas nécessaires pour modifier les conditions de son insertion dans la liste communautaire des substances actives.
Conditions des essais.
Les mêmes conditions que celles prévues à l'annexe I, point 6.5, sont applicables.
8.5. Résidus contenus dans les cultures suivantes.
But de l'essai.
L'objectif des présentes études est de permettre une évaluation des résidus pouvant être contenus dans les cultures suivantes.
Situations dans lesquelles les essais sont requis.
Des études complémentaires ne sont requises que s'il est impossible de procéder à une extrapolation à partir des données obtenues pour la substance active conformément aux dispositions de l'annexe I, point 6.6. Tel peut être le cas pour des formules spéciales, pour méthodes spéciales d'application ou pour des produits végétaux au sujet desquels des données n'avaient pas été présentées dans le cadre de l'insertion de la substance active ou n'étaient pas nécessaires pour modifier les conditions de son insertion dans la liste communautaire des substances actives.
Conditions des essais.
Les mêmes conditions que celles prévues à l'annexe I, point 6.6, sont applicables.
8.6. Limites maximales de résidus proposées et définition d'un résidu.
Les limites maximales de résidus proposées doivent être totalement justifiées et comprendre, le cas échéant, des données complètes relatives à l'analyse statistique appliquée.
Si les études sur le métabolisme présentées conformément aux dispositions du point 8.1 indiquent que la définition d'un résidu devrait être modifiée compte tenu de la définition actuelle d'un résidu et de l'évaluation nécessaire définies à l'annexe I, point 6.7, une réévaluation de la substance active peut être nécessaire.
8.7. Propositions relatives aux délais d'attente avant récolte pour les utilisations envisagées ou aux délais de rétention ou de stockage en cas d'utilisations postérieures à la récolte.
Les propositions doivent être entièrement justifiées.
8.8. Estimation de l'exposition potentielle ou réelle imputable au régime alimentaire ou à d'autres causes.
Il convient d'établir de manière réaliste la prévision de l'ingestion par le régime alimentaire ou fourrager, ce qui peut se faire de manière progressive et aboutir à une prévision de plus en plus réaliste de l'ingestion. S'il s'agit de facteurs importants, d'autres sources d'exposition, telles que les résidus provenant de l'utilisation de médicaments, notamment vétérinaires, doivent être prises en compte.
8.9. Résumé et évaluation du comportement des résidus.
Un résumé et une évaluation de toutes les données exposées dans la présente section doivent être effectués conformément aux lignes directrices établies par les autorités compétentes des Etats membres au sujet du format de tels résumés et évaluations. Le document doit comprendre une estimation détaillée et critique de ces données dans le contexte des lignes directrices et critères importants pour l'évaluation et la prise de décision, une importance particulière étant accordée aux risques éventuels ou réels pour l'homme et les animaux et à l'importance, la qualité et la fiabilité de la base de données. Si des données relatives au métabolisme ont été présentées, l'importance toxicologique de tout métabolite trouvé chez les animaux autres que les mammifères doit être examinée.
Un diagramme schématique doit être établi pour la voie métabolique dans les végétaux et animaux avec une brève explication de la distribution et des modifications chimiques en cause, si des données relatives au métabolisme ont été présentées.
9. Devenir et comportement dans l'environnement.
Introduction.
i) Les informations fournies, jointes à celles prévues à l'annexe I concernant la substance active, doivent être suffisantes pour permettre une évaluation du devenir et du comportement du produit phytopharmaceutique dans l'environnement, ainsi que des espèces non cibles susceptibles d'être menacées à la suite de l'exposition à ce produit.
ii) Les informations fournies relatives au produit phytopharmaceutique, jointes à d'autres informations pertinentes, et les informations fournies relatives à la substance active devront notamment être suffisantes pour :
- fixer les symboles de danger, les indications relatives au danger et les phrases types relatives à la nature des risques et aux conseils de prudence pour la protection de l'environnement à faire figurer sur l'emballage (conteneurs) ;
- prévoir la dispersion, le devenir et le comportement dans l'environnement ainsi que les durées correspondantes ;
- identifier les espèces et populations non cibles menacées à la suite d'une exposition potentielle,
et
- identifier les mesures nécessaires pour minimiser la contamination de l'environnement et l'impact sur les espèces non cibles.
iii) En cas d'utilisation de substances expérimentales radiomarquées, les dispositions de l'annexe I, section 7 :
"Introduction", point iv, sont applicables.
iv) Le cas échéant, les essais doivent être conçus et les données analysées en utilisant les méthodes statistiques adéquates.
Les analyses statistiques doivent être rapportées de manière exhaustive (par exemple, toutes les estimations ponctuelles doivent être rapportées avec les intervalles de confiance, les valeurs de probabilité exactes doivent être fournies plutôt que la mention "significatif/non significatif").
v) Concentrations prévisibles dans l'environnement dans le sol (CPES), dans l'eau (CPEesu et CPEgex) et dans l'air (CPEa).
Des estimations justifiées doivent être faites des concentrations prévisibles de la substance active et des métabolites et produits de dégradation et de réaction ayant une incidence dans le sol, les eaux souterraines, les eaux de surface et l'air, à la suite d'une utilisation actuelle ou proposée. De plus, une estimation correspondant au cas réaliste le plus défavorable doit être effectuée.
Pour l'estimation de ces concentrations, les définitions suivantes sont applicables.
Concentration prévisible dans l'environnement. Sol (CPES).
Le niveau de résidus dans la couche supérieure du sol auquel peuvent être exposés les organismes non cibles du sol (exposition aiguë et chronique).
Concentration prévisible dans l'environnement. Eaux de surface (CPEcsu).
Le niveau de résidus dans les eaux de surface auquel peuvent être exposés les organismes non cibles aquatiques (exposition aiguë et chronique).
Concentration prévisible dans l'environnement. Eaux souterraines (CPEcsu).
Le niveau de résidus dans les eaux souterraines.
Concentration prévisible dans l'environnement. Air (PECa).
Le niveau de résidus dans l'air auquel peuvent être exposés l'homme, les animaux et d'autres organismes non cibles (exposition aiguë et chronique).
Pour l'estimation de ces concentrations, il convient de tenir compte de toutes les informations pertinentes concernant le produit phytopharmaceutique et la substance active. Une approche utile pour ces estimations est fournie par les systèmes OEPP d'évaluation des risques environnementaux OEPP/EPPO (1993), "Systèmes de décision pour l'évaluation des effets non intentionnels des produits phytosanitaire sur l'environnement". Bulletins OEPP/EPPO n° 23, p. 1-154, et n° 24, p. 1-87.
Il conviendra, le cas échéant, d'utiliser les paramètres prévus au présent chapitre.
Quand des modèles sont utilisées pour l'estimation des concentrations prévisibles dans l'environnement, il doivent :
- fournir la meilleure appréciation possible de tous les processus pertinents mis en jeu en tenant compte de paramètres et hypothèses réalistes ;
- être dans la mesure du possible validés de manière fiable par des mesures effectuées dans des conditions pertinentes pour l'utilisation du modèle ;
- correspondre aux conditions de la zone d'utilisation.
Les informations fournies doivent si nécessaire comprendre les informations visées à l'annexe I, partie A, point 7.
9.1. Devenir et comportement dans le sol.
Le cas échéant, les mêmes dispositions concernant les informations à fournir sur le sol utilisé et sa sélection sont applicables selon les dispositions prévues à l'annexe I, point 7.1.
9.1.1. Vitesse de dégradation dans le sol.
9.1.1.1. Etudes de laboratoire.
But des essais.
Les études de dégradation dans le sol doivent fournir les meilleures estimations possibles du temps nécessaire à la dégradation de 50 p. 100 et de 90 p. 100 (DT50 lab. et DT90 lab.) de la substance active dans des conditions de laboratoire.
Situations dans lesquelles les essais sont requis.
La persistance et le comportement des produits phytopharmaceutiques dans le sol doivent être étudiés, sauf quand il est possible de les extrapoler à partir des données obtenues sur la substance active et les métabolites et produits de dégradations et de réaction ayant une incidence toxicologique et environnementale conformément aux exigences de l'annexe I, section 7, point 7.1.1.2. Ces extrapolations sont par exemple impossibles pour les préparations à libération lente.
Modalités des essais.
La vitesse de dégradation en conditions aérobies et/ou anaérobies dans le sol doit être rapportée. La durée normale de l'étude est de 120 jours, sauf si plus de 90 p. 100 de la substance active sont dégradés avant l'expiration de cette période.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'ecotoxicité des pesticides.
9.1.1.2. Etudes de terrain.
Etudes de dissipation dans le sol.
But des essais.
Les études de dissipation dans le sol doivent fournir les meilleures estimations possibles du temps nécessaire à la dissipation de 50 p. 100 et de 90 p. 100 (DT50f et DT90f) de la substance active dans des conditions de terrain. Le cas échéant, des informations concernant les métabolites et les produits de dégradation et de réaction ayant une incidence toxicologique et environnementale doivent être rapportées.
Situations dans lesquelles les essais sont requis.
La dissipation et le comportement des produits phytopharmaceutiques dans le sol doivent être étudiés, sauf s'il est possible de les extrapoler à partir des données obtenues sur la substance active et les produits de dégradation et de réaction et métabolites ayant une incidence toxicologique et environnementale conformément aux exigences de l'annexe I, section 7, point 7.1.1.2. Cette extrapolation est par exemple impossible pour les préparations à libération lente.
Modalités et ligne directrice des essais.
Mêmes dispositions qu'au titre correspondant de l'annexe I, section 7, point 7.1.1.2.2.
Etudes des résidus dans le sol.
But des essais.
Les études de résidus dans le sol doivent fournir des estimations des niveaux de résidus dans le sol au moment de la récolte, ou au moment des semis ou de la mise en place des cultures suivantes.
Situations dans lesquelles les essais sont requis.
Les études de résidus dans le sol doivent être rapportées sauf s'il est possible d'extrapoler les résultats à partir des données obtenues sur la substance active et les métabolites et produits de dégradation et de réaction ayant une incidence significative conformément aux exigences de l'annexe I, section 7, point 7.1.1.2.2. Cette extrapolation est par exemple impossible pour les préparations à libération lente.
Modalités des essais.
Mêmes dispositions qu'au titre correspondant de l'annexe I, section 7, point 7.1.1.2.2.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
Etudes d'accumulation dans le sol.
But des essais.
Les essais doivent fournir des données suffisantes permettant d'évaluer la possibilité d'accumulation des résidus de la substance active et des produits de réaction et de dégradation, ainsi que des métabolites ayant une incidence toxicologique et environnementale.
Situations dans lesquelles les essais sont requis.
Des études d'accumulation dans le sol doivent être rapportées sauf s'il est possible d'extrapoler les résultats à partir de données obtenues sur la substance active et les métabolites et produits de dégradation et de réaction ayant une incidence environnementale conformément aux exigences de l'annexe I, section 7, point 7.1.1.2.2. Ces extrapolations sont par exemple impossibles pour les préparations à libération lente.
Modalités des essais.
Mêmes dispositions qu'au titre correspondant de l'annexe I, section 7, point 7.1.1.2.2.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
9.1.2. Mobilité dans le sol.
But des essais.
Les essais doivent fournir des données suffisantes permettant d'évaluer le potentiel de mobilité et de lixiviation de la substance active et des métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale.
9.1.2.1. Etudes de laboratoire.
Situations dans lesquelles les essais sont requis.
La mobilité des produits phytopharmaceutiques dans le sol doit être étudiée, sauf s'il est possible d'extrapoler les résultats à partir de données obtenues conformément aux exigences de l'annexe I, section 7, points 7.1.2 et 7.1.3. Cette extrapolation est par exemple impossible pour les préparations à libération lente.
Ligne directrice des essais.
Méthodes Setac d'évaluation du devenir dans l'environnement et de l'écotoxicité des pesticides.
9.1.2.2. Etudes lysimétriques ou études de lixiviation sur le terrain.
But des essais.
L'essai doit fournir des données concernant :
- la mobilité du produit phytopharmaceutique dans le sol ;
- le potentiel de lixiviation vers les eaux souterraines ;
- la dispersion potentielle dans les sols.
Situations dans lesquelles les essais sont requis.
L'avis de spécialistes sera nécessaire pour déterminer si des études de lixiviation sur le terrain ou des études lysimétriques doivent être effectuées, compte tenu des résultats des études de dégradation et de mobilité et de la CPEs calculée. Le type d'étude à effectuer doit faire l'objet d'une discussion avec les autorités compétentes.
Ces études doivent être effectuées sauf s'il est possible d'extrapoler les résultats à partir de données obtenues sur la substance active et les métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale conformément aux exigences de l'annexe I, section 7, point 7.1.3. Cette extrapolation est par exemple impossible pour les préparations à libération lente.
Modalités des essais.
Mêmes dispositions qu'au titre correspondant de l'annexe I, section 7, point 7.1.3.3.
9.1.3. Estimation des concentrations prévisibles dans le sol.
Les estimations des CPE, doivent correspondre à la fois à une application unique à la dose d'application la plus élevée pour laquelle une autorisation est demandée et au nombre maximal d'applications à la dose la plus élevée pour lesquels l'autorisation est demandée, pour chaque sol testé pertinent ; elles sont exprimées en milligrammes de substance active et de métabolites et produits de dégradation et de réaction ayant une incidence environnementale par kilogramme de sol. Les facteurs à prendre en considération lors des estimations de CPE, concernent l'application directe et indirecte au sol, l'entraînement, le ruissellement et la lixiviation, et comprennent des processus tels que la volatilisation, l'adsorption, l'hydrolyse, la photolyse, la dégradation aérobie et anaérobie. Dans le calcul de la CPE, on peut utiliser d'une densité apparente des sols de 1,5 g/cm3 de poids sec, une profondeur de couche de sol de 5 cm pour les applications de surface et de 20 cm en cas d'incorporation dans le sol. En cas de présence d'une couverture végétale au moment de l'application, on peut supposer que 50 p. 100 (au minimum) de la dose appliquée atteignent la surface du sol sous réserve d'informations plus spécifiques fournies par des données expérimentales.
Il convient de fournir des estimations de la CPE, initiales à court terme et à long terme (moyennes pondérées dans le temps) :
- initiales : immédiatement après l'application ;
- à court terme : 24 heures, 2 jours et 4 jours après la dernière application ;
- à long terme : 28, 50 et 100 jours après la dernière application, selon le cas.
9.2. Devenir et comportement dans l'eau.
9.2.1. Estimation des concentrations dans les eaux souterraines.
Les voies de contamination des eaux souterraines doivent être définies en tenant compte des conditions phytosanitaires, agronomiques et environnementales pertinentes (y compris climatiques).
Des estimations (calculs) appropriées de la concentration prévisible dans les eaux souterraines CPEGw de la substance active et des métabolites et produits de dégradation et de réaction ayant une incidence doivent être fournies.
Les estimations de la CPE doivent correspondre au nombre maximal et aux doses les plus élevées d'application pour lesquels une autorisation est demandée.
L'avis de spécialistes est requis afin de déterminer si des essais de terrain supplémentaires pourraient fournir des informations utiles. Avant d'effectuer ces études, le demandeur doit demander l'accord des autorités compétentes en ce qui concerne le type d'étude à effectuer.
9.2.2. Impact sur les méthodes de traitement des eaux.
Dans les cas où ces informations sont nécessaires dans le cadre d'une autorisation conditionnelle visée à l'annexe VI, partie C, point 2.5.1.2. b), les informations fournies devront permettre d'établir ou d'estimer l'efficacité des méthodes de traitement des eaux (eau potable et eaux usées) et l'impact sur ces méthodes. Avant d'effectuer des études, le pétitionnaire doit obtenir l'accord des autorités compétentes sur le type d'informations à fournir.
9.2.3. Estimation des concentrations dans les eaux de surface.
Les voies de contamination des eaux de surface doivent être définies en tenant compte des conditions agronomiques, phytosanitaires et environnementales (y compris climatiques) pertinentes. Des estimations (calculs) appropriées de la concentration environnementale prévisible dans les eaux de surface CEP5w de la substance active et des métabolites et produits de dégradation et de réaction ayant une incidence toxicologique et environnementale doivent être fournies.
Les estimations de la CEP doivent correspondre au nombre maximal et aux doses les plus élevées d'application pour lesquels l'autorisation est demandée et concerner les lacs, les étangs, les rivières, les canaux, les fleuves, les canaux d'irrigation ou de drainage et les drains.
Les facteurs à prendre en compte lors des estimations de la CEP5w concernent l'application directe à l'eau, la dérive de pulvérisation, le ruissellement, la décharge par les drains et le dépôt atmosphérique, et comprennent des processus tels que la volatilisation, l'adsorption, l'advection, l'hydrolyse, la photolyse, la biodégradation, la sédimentation et la remise en suspension.
Des calculs initiaux à court terme et à long terme CEP5w concernant les masses d'eau stagnantes et à écoulement lent (moyennes pondérées dans le temps) doivent être fournis :
- initiaux : immédiatement après l'application ;
- à court terme : 24 heures, 2 jours et 4 jours après la dernière application ;
- à long terme : 7, 14, 21, 28 et 42 jours après la dernière application, selon le cas.
L'avis de spécialistes est requis afin de déterminer si des essais de terrain supplémentaires pourraient fournir des informations utiles. Avant d'effectuer ces études, le demandeur doit obtenir l'accord des autorités compétentes sur le type d'étude à effectuer.
9.3. Sort et comportement dans l'air.
Instructions en cours d'élaboration.
Article Annexe II En savoir plus sur ct article...
Modifié par Arrêté 1996-02-01 art. 2 JORF 20 février 1996
Modifié par Arrêté 1996-09-24 art. 6 JORF 27 octobre 1996
10. Etudes écotoxicologiques.
Introduction.
i) Les informations fournies, jointes à celles qui concernent la (les) substance(s) actives(s), doivent être suffisantes pour permettre d'évaluer l'impact sur les espèces non cibles (flore et faune) du produit phytopharmaceutique, utilisé selon les instructions d'emploi indiquées. L'impact peut être dû à une exposition unique, prolongée ou répétée, et peut être réversible ou irréversible.
ii) En particulier, les informations relatives au produit phytopharmaceutique et les autres données pertinentes, ainsi que les informations relatives à la substance active doivent être suffisantes pour :
- définir les symboles des dangers, les indications du danger et les phrases relatives à la nature des risques ainsi que les conseils de prudence pour la protection de l'environnement à faire figurer sur les emballages (conteneurs) ;
- permettre une évaluation des risques à court aussi bien qu'à long terme pour les espèces non cibles (populations, communautés et processus, selon les cas) ;
- permettre de décider si des précautions particulières doivent être prises pour protéger les espèces non cibles.
iii) Il est nécessaire de faire état de tous les effets potentiellement nuisibles constatés au cours des investigations écotoxicologiques de routine et de réaliser et rapporter les études supplémentaires qui se révéleraient nécessaires pour les mécanismes en cause et évaluer la signification de ces effets.
iv) D'une façon générale, un grand nombre des données relatives à l'impact sur les espèces non cibles requises pour l'autorisation du produit phytopharmaceutique auront été soumises et évaluées aux fins de l'inclusion de(s) la substance(s) active(s) dans la liste. Les informations sur le sort et le comportement dans l'environnement, établies et présentées conformément au chapitre 9, points 9.1 à 9.3, et sur les niveaux de résidus dans les végétaux fournis et présentés conformément au chapitre 8 sont essentielles pour l'évaluation de l'impact sur les espèces non cibles, car elles fournissent des informations sur la nature et l'ampleur de l'exposition potentielle ou réelle. Les estimations relatives à la PEC finale doivent être adaptées en fonction des divers groupes ou organismes en tenant compte, en particulier, de la biologie des espèces les plus sensibles.
Les études et les informations toxicologiques présentées conformément au chapitre 7, point 7.1, fournissent des données essentielles sur la toxicité pour les vertébrés.
v) S'il y a lieu, des essais seront mis au point et les données obtenues seront analysées à l'aide de méthodes statistiques appropriées. Tous les détails de l'analyse statistique sont à noter (par exemple, toutes les estimations doivent être délimitées par un intervalle de confiance, il vaut mieux indiquer des valeurs "p" exactes au lieu de préciser qu'une valeur est significative/non significative).
vi) Si une étude comporte l'utilisation de doses différentes, la relation entre dose et effet néfaste doit être notée.
vii) S'il est nécessaire de disposer des facteurs de l'exposition pour décider si une étude doit être effectuée, il y a lieu d'utiliser les données obtenues conformément aux dispositions de l'annexe II, chapitre 9.
Toutes les données utiles concernant le produit phytopharmaceutique et la substance active doivent être prises en considération pour l'évaluation de l'exposition. Les programmes d'évaluation du risque environnemental de l'OEPPOEPP/EPPO (1993). Programmes décisionnels d'évaluation du risque environnemental des produits phytosanitaires. Bulletins OEPP/EPPO n° 23, p. 1-154, et n° 24, p. 1-87, constituent une approche utile pour ces évaluations. Si nécessaire, on utilisera les paramètres exposés dans le présent chapitre.
Si les données disponibles indiquent que le produit phytopharmaceutique est plus toxique que la substance active, les données relatives à la toxicité du produit phytopharmaceutique doivent être utilisées pour le calcul des rapports toxicité/exposition importants.
viii) Dans le contexte de l'influence que les impuretés peuvent exercer sur le comportement écotoxicologique, il est indispensable de fournir, pour chacune des études soumises, une description détaillée (spécifications) du matériel utilisé, conformément aux prescriptions du chapitre 1, point 4.
ix) Pour faciliter l'évaluation de la signification des résultats obtenus, il y a lieu d'utiliser, dans la mesure du possible, la même souche de l'espèce concernée pour les différents essais de toxicité.
10.1. Effets sur les oiseaux.
Les effets que la substance peut avoir sur les oiseaux doivent être étudiés, sauf lorsque l'éventualité d'une exposition directe ou indirecte des oiseaux peut être exclue, comme, par exemple, en cas d'utilisation en espace clos ou pour le traitement des plaies et blessures.
Le rapport toxicité aiguë/exposition (TERa) et le rapport toxicité alimentaire à court terme/exposition (TERst) et le rapport toxicité alimentaire à long terme/exposition (TERlt) doivent être établis, étant entendu que :
TERa = DL 50 (mg de s.a./kg de poids corporel)/ETE (mg de s.a./kg de poids corporel) ;
TERst = CL 50 (mg de s.a./kg d'aliment)/ETE (mg de s.a./kg d'aliment) ;
TERlt = CSEO (mg de s.a./kg d'aliment)/ETE (mg de s.a./kg d'aliment),
où ETE = exposition théorique estimée.
Dans le cas des pastilles, granules ou graines traitées, il y a lieu de noter la concentration de s.a. de chaque pastille, granule ou graine ainsi que la proportion de DL 50 pour la s.a. dans 100 particules et par gramme de particule. La taille et la forme des pastilles ou granules doivent être spécifiées.
Dans le cas des appâts, la concentration de s.a. dans l'appât (mg/kg) doit être précisée.
10.1.1. Toxicité orale aiguë.
Objet de l'essai.
Dans la mesure du possible, l'essai devrait permettre d'établir les valeurs de DL 50, la dose seuil létale, les temps de réponse et de récupération et le NSEO et doit faire état des observations pathologiques significatives à l'autopsie.
Circonstances où l'essai est requis.
La toxicité orale aiguë des préparations doit être établie lorsque TERg ou TERst de la (des) substance(s) active(s) chez les oiseaux est compris entre 10 et 100 ou lorsque les résultats des essais sur les mammifères montrent une toxicité significativement plus élevée de la préparation par rapport à la substance active, sauf s'il est prouvé qu'une exposition des oiseaux au produit phytopharmaceutique proprement dit est improbable.
Conditions de l'essai.
L'étude doit être effectuée sur l'espèce la plus sensible déterminée dans les études visées à l'annexe I, points 8.1.1 ou 8.1.2.
10.1.2. Essais supervisés en cage ou sur le terrain.
Objet de l'essai.
L'essai doit permettre d'obtenir des données suffisantes pour évaluer la nature et l'ampleur du risque dans les conditions pratiques d'utilisation.
Circonstances où l'essai est requis.
Lorsque TERa et TERst 100 et lorsque d'autres études sur la substance active (par exemple, des études de reproduction) n'ont pas révélé l'existence de risques, il n'est pas nécessaire de procéder à d'autres essais. Dans les autres cas, un jugement d'expert est requis pour décider s'il est nécessaire de procéder à des études plus approfondies. L'avis spécialisé tiendra compte, selon les cas, du comportement alimentaire, de la répulsivité, des aliments de remplacement, de la teneur effective en résidus de l'aliment, de la persistance du composé dans la végétation, de la dégradation du produit formulé ou des denrées traitées, de la part de prédation dans la nourriture de l'acceptation de l'appât, des granules ou des graines traitées et de la possibilité d'une bioconcentration.
Lorsque TERa et TERst sont 10 ou que TERlt est 5, il est indispensable d'effectuer des essais en cage ou sur le terrain et d'en rapporter les résultats à moins qu'une évaluation finale ne soit possible sur la base d'études effectuées conformément au point 10.1.3.
Conditions de l'essai.
Avant de réaliser ces études, le demandeur doit solliciter l'agrément des autorités compétentes sur la nature et les conditions de l'étude à réaliser.
10.1.3. Appétence pour les oiseaux des appâts, des granules ou des semences traitées.
Objet de l'essai.
L'essai doit permettre d'obtenir des données suffisantes pour évaluer la possibilité qu'a le produit phytopharmaceutique ou le produit végétal auquel celui-ci a été appliqué d'être consommé par les oiseaux.
Circonstances où l'essai est requis.
Les essais d'appétence (palatabilité) doivent être réalisés dans le cas des semences traitées, des pastilles et des appâts ainsi que de préparations en granules et lorsque TERa 10.
10.1.4. Effets d'empoisonnement secondaire.
Un jugement d'expert est requis pour décider de la nécessité de procéder ou non à une étude des effets d'empoisonnement secondaire.
10.2. Effets sur les organismes aquatiques.
Les effets que la substance peut avoir sur les espèces aquatiques doivent être étudiés, sauf lorsque l'éventualité d'une exposition de ces espèces peut être exclue.
Il y a lieu de déterminer TERa et TERlt, sachant que :
TERa = CL 50 aiguë (mg de s.a./l)/CEPsw dans l'hypothèse réaliste la plus défavorable, initiale ou à court terme (mg de s.a./l) ;
TERlt = CSEO chronique (mg de s.a./l)/CEPsw long terme (mg de s.a./l) ;
10.2.1. Toxicité aiguë pour les poissons, les invertébrés aquatiques ou effets sur la croissance des algues.
Circonstances où l'essai est requis.
En principe, l'essai devrait être effectué sur une des espèces de chacun des trois groupes d'organismes aquatiques visés à l'annexe II, point 8.2 (poissons, invertébrés aquatiques et algues), lorsque le produit phytopharmaceutique lui-même peut contaminer l'eau. Toutefois, lorsque les informations disponibles permettent de conclure qu'un de ces groupes est nettement plus sensible, les essais ne doivent être effectués que sur l'espèce la plus sensible du groupe en cause.
Il y a lieu de réaliser l'essai :
- lorsque la toxicité aiguë du produit phytopharmaceutique ne peut pas être prédite sur la base des données relatives à la substance active, ce qui est notamment le cas lorsque la formulation comporte deux ou plusieurs ingrédients ou substances actives tels que des solvants, des émulgateurs, des agents tensioactifs, des dispersants et des engrais pouvant accroître la toxicité par rapport à la substance active,
ou
- lorsque l'utilisation envisagée prévoit une application directe sur l'eau,
à moins que des études appropriées visées au point 10.2.4 ne soient disponibles.
Conditions de l'essai et lignes directrices.
Les dispositions pertinentes prévues aux points correspondants de l'annexe I, chapitre 8, points 8.2.1, 8.2.4 et 8.2.6 sont applicables.
10.2.2. Etudes de microcosme ou de mésocosme.
Objet de l'essai.
L'essai doit permettre d'obtenir des données suffisantes pour évaluer l'impact essentiel sur les organismes aquatiques dans les conditions de la pratique.
Circonstances où l'essai est requis.
Si TERa ou si TERlt 10, un jugement d'expert est requis pour décider s'il y a lieu ou non de procéder à une étude de microcosme ou de mésocosme. Cet avis doit tenir compte de toute donnée disponible en sus de celles requises par les dispositions de l'annexe I, chapitre 8, points 8.2 et du point 10.2.1.
Conditions de l'essai.
Avant de réaliser ces études, le demandeur doit solliciter l'agrément des autorités compétentes sur les objectifs spécifiques et, par conséquent, sur la nature et les conditions de l'étude à réaliser.
L'étude doit porter au moins sur le taux maximal d'exposition probable, qu'il résulte de l'application directe, de la dérive, du drainage ou du ruissellement. La durée de l'étude doit être suffisante pour permettre l'évaluation de tous les effets.
Lignes directrices.
Les lignes directrices adéquates sont contenues dans :
- les lignes directrices du Setac sur les procédures d'essai des pesticides dans les mésocosmes d'eau douce/Atelier Huntingdon, 3 et 4 juillet 1991,
ou
- les essais naturels en eau douce pour l'évaluation des risques des produits chimiques - European Workshop on Freshwater Field Tests (EWOFFT).
10.2.3. Données sur les résidus dans les poissons.
Objet de l'essai.
L'essai doit permettre d'obtenir des données suffisantes pour évaluer la présence potentielle de résidus dans les poissons.
Circonstances où l'essai est requis.
D'une façon générale, des données peuvent être obtenues à partir des études de bioconcentration chez le poisson.
Si une bioconcentration a été observée dans l'étude réalisée conformément à l'annexe I, chapitre 8, point 8.2.3, un jugement d'expert est requis pour décider de la nécessité de procéder ou non à une étude à long terme de microcosme ou de mésocosme en vue d'établir la quantité maximale de résidus risquant d'être constatée.
Lignes directrices.
Lignes directrices du Setac sur les procédures d'essai des pesticides dans les mésocosmes d'eau douce-Atelier Huntingdon, 3 et 4 juillet 1991.
10.2.4. Etudes supplémentaires.
Les études visées à l'annexe I, points 8.2.2 et 8.2.5, peuvent se révéler nécessaires pour des produits phytopharmaceutiques particuliers (...), lorsqu'il n'est pas possible de procéder à une extrapolation des données obtenues dans les études correspondantes sur la substance active.
10.3. Effets sur les vertébrés terrestres autres que les oiseaux.
Les effets éventuels sur les vertébrés sauvages doivent être étudiés, sauf lorsque l'éventualité d'une exposition directe ou indirecte des vertébrés terrestres autres que les oiseaux peut être exclue. Il y a lieu de déterminer TERa et TERst et TERlt sachant que :
TERa = DL 50 (mg de s.a./kg de poids corporel)/ETE (mg de s.a./kg de poids corporel) ;
TERst = NSEO subchronique (mg de s.a./kg d'aliment)/ETE (mg de s.a./kg d'aliment) ;
TERlt = NSEO chronique (mg de s.a./kg d'aliment)/ETE (mg de s.a./kg d'aliment), ETE étant l'exposition théorique estimée.
En principe, l'ordre des opérations pour l'évaluation de risques pour ces espèces est similaire à celui qui est prévu pour les oiseaux. Dans la pratique, il est souvent superflu de procéder aux études additionnelles, car les études menées conformément aux exigences de l'annexe I, chapitre 5, et de l'annexe II, chapitre 7, permettent de déduire les informations requises.
Objet de l'essai.
L'essai doit permettre d'obtenir des informations suffisantes pour évaluer la nature et l'ampleur des risques pour les vertébrés terrestres autres que les oiseaux dans les conditions d'utilisation pratique.
Circonstances où l'essai est requis.
Lorsque TERa et TERst 100 et lorsque d'autres études font apparaître l'absence de risques supplémentaires, il n'est pas nécessaire de continuer les essais. Dans les autres cas, un jugement d'expert est requis pour décider s'il est nécessaire de procéder à des études plus approfondies. L'avis spécialisé tiendra compte, selon les cas, du comportement alimentaire, de la répulsivité, des aliments de remplacement, de la teneur effective en résidus de l'aliment, de la persistance du composé dans la végétation, de la dégradation du produit formulé ou des denrées traitées, de la part de prédation dans la nourriture, de l'acceptation alimentaire de l'appât, des granules ou des graines traitées et de la possibilité d'une bioconcentration.
Lorsque TERa et TERst sont 10 ou que TERlt est 5, il y a lieu d'effectuer des essais en cage ou d'autres études appropriées.
Conditions de l'essai.
Avant de réaliser ces études, le demandeur doit solliciter l'agrément des autorités compétentes sur la nature et les conditions de l'étude à réaliser et sur la détermination ou non des effets d'empoisonnement secondaire.
10.4. Effets sur les abeilles.
Les effets sur les abeilles doivent être étudiés, sauf lorsque le produit est destiné à être utilisé exclusivement dans des situations où l'exposition des abeilles est improbable, telles que :
- l'entreposage de denrées alimentaires en espace clos ;
- le traitement non systémique des semences ;
- les préparations non systémiques pour l'épandage sur le sol ;
- les traitements non systémiques par trempage des bulbes et plants repiqués ;
- les traitements des plaies et des blessures ;
- les apâts rodenticides ;
- l'emploi en serre sans pollinisateurs.
Il y a lieu de déterminer les quotients de risque concernant l'exposition orale ou de contact (QHO et QHC) :
QHO = Dose/DL 50 orale (m de s.a. par abeille) ;
QHC = Dose/DL 50 de contact (m de s.a. par abeille),
la dose étant la dose maximale d'application pour laquelle l'autorisation est demandée, exprimée en grammes de substance active par hectare.
10.4.1. Toxicité aiguë orale ou de contact.
Objet de l'essai.
L'essai devrait permettre d'établir les valeurs de la DL 50 (à la suite d'une exposition orale ou de contact).
Circonstances où l'essai est requis.
L'essai est requis si :
- le produit contient plus d'une substance active ;
- on ne peut prédire avec une fiabilité suffisante que la toxicité d'une nouvelle formulation sera égale ou inférieure à celle d'une formulation testée selon les dispositions de l'annexe I, chapitre 8, point 8.3.1.1, ou du présent point.
Lignes directrices.
L'essai devrait être effectué en conformité avec la directive 170 de l'OEPP.
10.4.2. Détermination des résidus.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer les risques éventuels que les traces résiduelles de produits phytopharmaceutiques restant sur les cultures présentent pour les abeilles butineuses.
Circonstances où l'essai est requis.
Lorsque QHC 50, un jugement d'expert est nécessaire pour décider s'il y a lieu de déterminer l'effet des résidus, sauf s'il est établi qu'aucune trace résiduelle significative de nature à affecter les abeilles butineuses ne subsiste sur les cultures ou si les essais en cage, en tunnel ou sur le terrain ont fourni des informations suffisantes.
Conditions de l'essai.
Il est indispensable de déterminer le temps létal 50 p. 100 (TL 50) (exprimé en heures) après 24 heures d'exposition aux résidus sur des feuilles vieillies pendant 8 heures. Si le TL 50 dépasse 8 heures, il n'y a pas lieu de procéder à d'autres essais.
10.4.3. Essais en cage.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer les risques éventuels que présente le produit phytopharmaceutique pour la survie et le comportement des abeilles.
Circonstances où l'essai est requis.
Lorsque QHO et QHC sont 50, il n'y a pas lieu de procéder à des essais supplémentaires, sauf si l'essai d'alimentation du couvain d'abeilles révèle des effets significatifs ou s'il existe des indices d'effets indirects telle une action à retardement ou une modification du comportement des abeilles ; dans ces cas, il y a lieu de pratiquer des essais en cage ou de terrain.
Il y a lieu de pratiquer des essais en cage ou de terrain lorsque QHO et QHC sont 50.
Si l'essai sur le terrain a été pratiqué et consigné conformément au point 10.4.4, il n'est pas nécessaire de procéder à un essai en cage. Toutefois, si un essai en cage est effectué, il doit être mentionné.
Conditions de l'essai.
L'essai devrait être pratiqué sur des abeilles saines. Si les abeilles ont été traitées, par exemple avec un varroacide, il y a lieu d'attendre quatre semaines avant d'utiliser l'essaim.
Lignes directrices.
L'essai doit être réalisé en conformité avec la directive 170 de l'OEPP.
10.4.4. Essais de terrain.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer les risques que le produit phytopharmaceutique peut présenter pour le comportement des abeilles et pour la survie et le développement de l'essaim.
Circonstances où l'essai est requis.
Les essais de terrain doivent être pratiqués lorsque l'essai en cage révèle des effets jugés significatifs par un spécialiste, compte tenu de l'utilisation envisagée, du sort et du comportement de la substance active.
Conditions de l'essai.
Les essais doivent être pratiqués sur des essaims d'abeilles mellifères saines présentant une vigueur naturelle identique. Si les abeilles ont été traitées par exemple au varroacide, il y a lieu d'attendre quatre semaines avant d'utiliser l'essaim. Les essais doivent être pratiqués dans des conditions dûment représentatives de l'utilisation envisagée.
Des effets particuliers (toxicité pour les larves, effets résiduels de longue durée, effets désorientants pour les abeilles) apparus lors des essais de terrain peuvent nécessiter d'autres investigations utilisant des méthodes spécifiques.
Lignes directrices.
Les essais doivent être réalisés en conformité avec la directive 170 de l'OEPP.
10.4.5. Essais en tunnel.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer l'impact que le butinage de fleurs ou de miellat contaminés exerce sur les abeilles.
Circonstances où l'essai est requis.
Lorsque les essais en cage ou de terrain ne permettent pas d'étudier certains effets, il y a lieu d'effectuer un essai en tunnel ; par exemple, lorsqu'il s'agit de produits phytopharmaceutiques destinés à lutter contre les pucerons et d'autres insectes suceurs.
Conditions de l'essai.
L'essai devrait être pratiqué sur des abeilles saines. Si les abeilles ont été traitées par exemple à l'aide d'un varroacide, il y a lieu d'attendre quatre semaines avant d'utiliser l'essaim.
Lignes directrices.
L'essai doit être réalisé en conformité avec la directive 170 de l'OEPP.
10.5. Effets sur les arthropodes autres que les abeilles.
Il y a lieu de déterminer les effets des produits phytopharmaceutiques sur les arthropodes terrestres non cibles (par exemple, les prédateurs ou les parasitoïdes des organismes nuisibles). Les renseignements obtenus sur ces espèces peuvent aussi être utilisés pour indiquer la toxicité potentielle pour d'autres espèces non cibles vivant dans le même environnement.
10.5.1. Essais au niveau du laboratoire, en condition semi-naturelle.
Objet de l'essai.
L'essai devrait permettre d'obtenir des informations suffisantes pour évaluer la toxicité du produit phytopharmaceutique pour certaines espèces d'arthropodes concernées par l'utilisation envisagée du produit.
Circonstances où l'essai est requis.
Il n'est pas nécessaire d'effectuer l'essai lorsque les données disponibles pertinentes permettent de prédire une forte toxicité (> 99 p. 100 d'effets sur les organismes par comparaison avec le témoin) ou lorsque le produit phytopharmaceutique est destiné à être utilisé exclusivement dans des situations où les arthropodes non cibles ne sont pas exposés, telles que :
- l'entreposage de denrées alimentaires en espace clos ;
- les traitements de plaies et blessures ;
- les appâts rodenticides.
L'essai doit être effectué lorsque les essais de laboratoire, pratiqués selon les exigences de l'annexe I, chapitre 8, point 8.3.2, et à la dose recommandée maximale, font apparaître des effets significatifs sur les organismes par comparaison avec le témoin. Les effets sur une espèce particulière sont considérés comme significatifs lorsqu'ils dépassent les valeurs de seuil définies dans les programmes d'évaluation des risques écologiques de l'OEPP à moins que des valeurs de seuil spécifiques ne soient définies dans les directives considérées.
L'essai est également requis lorsque :
- le produit contient plus d'une substance active ;
- on ne peut prédire avec une fiabilité suffisante que la toxicité d'une nouvelle formulation sera égale ou inférieure à celle d'une formulation testée selon les dispositions de l'annexe I, chapitre 8, point 8.3.2, ou du présent point ;
- le mode d'emploi proposé ou le sort et le comportement du produit font prévoir une exposition constante ou répétée ;
- l'utilisation proposée fait l'objet d'un changement significatif, par exemple, en passant des grandes cultures aux vergers, et que les espèces concernées par la nouvelle utilisation n'ont pas été testées au préalable ;
- la dose d'application recommandée est relevée au-delà du niveau précédemment testé conformément aux dispositions de l'annexe I.
Conditions de l'essai.
Lorsque des effets significatifs ont été observés dans les études réalisées conformément aux exigences de l'annexe I, chapitre 8, point 8.3.2, ou en cas de changement d'utilisation tel que le passage des grandes cultures aux vergers, la toxicité à l'égard de deux espèces concernées supplémentaires doit être étudiée et rapportée. Ces espèces doivent différer des espèces concernées déjà testées conformément à l'annexe I, chapitre 8, point 8.3.2.
Dans le cas d'un changement composé ou d'une nouvelle formulation, la toxicité sera évaluée, dans un premier temps, à l'aide des deux espèces les plus sensibles identifiées par les études déjà réalisées et pour lesquelles les valeurs de seuil ont été dépassées sans que les effets excèdent 99 p. 100. Il sera ainsi possible de procéder à une comparaison : si la toxicité apparaît nettement plus élevée, il faut tester deux espèces appropriées à l'utilisation proposée du produit.
L'essai doit être effectué en utilisant une dose équivalente à la dose maximale d'application pour laquelle l'autorisation est demandée. Il convient d'adopter une démarche d'essai progressive qui commence par le laboratoire avant de passer, si nécessaire, en condition semi-naturelle.
Si le produit doit être appliqué plus d'une fois par saison, il y a lieu d'utiliser le double de la dose d'application recommandée, sauf si cette information ressort déjà des études réalisées conformément à l'annexe II, chapitre 8, point 8.3.2.
Lorsque le mode d'emploi proposé ou le sort et le comportement du produit font prévoir une exposition continue ou répétée (par exemple, si le produit doit être appliqué plus de trois fois par saison, l'intervalle entre deux applications étant de 15 jours ou moins), il appartient à un expert d'établir si des essais supplémentaires s'imposent en sus des essais initiaux de laboratoire afin de refléter l'utilisation proposée. Ces essais peuvent être réalisés en laboratoire ou dans des conditions semi-naturelles. S'ils sont effectués en laboratoire, il convient d'utiliser un substrat réaliste tel que des matériels végétaux ou un sol naturel. Il peut cependant se révéler plus approprié de procéder à des essais de terrain.
Lignes directrices.
Les essais qui s'imposent doivent être réalisés en conformité avec les lignes directrices appropriées qui répondent au moins aux conditions d'essai prévues par le Setac, Guidance Document on Regulatory Testing Procedures for Pesticides with Non-Target Arthropods.
10.5.2. Essais sur le terrain.
Objet de l'essai.
Les essais devraient permettre d'obtenir des informations suffisantes pour évaluer le risque que présente le produit phytopharmaceutique pour les arthropodes dans les conditions réelles.
Circonstances où l'essai est requis.
En cas d'observation d'effets significatifs après exposition en laboratoire ou dans des conditions semi-naturelles ou lorsque le mode d'emploi proposé ou le sort et le comportement du produit font prévoir une exposition continue ou répétée, un jugement d'expert sera nécessaire pour établir s'il y a lieu de procéder à des essais plus approfondis pour obtenir une évaluation exacte du risque.
Conditions de l'essai.
Les essais doivent être réalisés dans des conditions représentatives de la réalité agricole et en conformité avec les recommandations d'utilisation proposées, de manière à produire une étude réaliste du cas le plus défavorable.
Tous les essais doivent comporter un étalon toxique.
Lignes directrices.
Les essais qui s'imposent doivent être réalisés en conformité avec les lignes directrices appropriées du Setac, Guidance Document on Regulatory Testing Procedures for Pesticides with Non-Target Arthropods.
10.6. Effets sur les vers de terre et d'autres macro-organismes non cibles du sol supposés être exposés à un risque.
10.6.1. Effets sur les vers de terre.
Il y a lieu de déterminer les effets possibles sur les vers de terre, sauf s'il est établi que leur exposition directe ou indirecte est improbable.
Il y a lieu de déterminer TERa et TER, sachant que :
TERa = CL 50 (mg s.a./kg)/CEPs dans l'hypothèse réaliste la plus défavorable, initiale ou à court terme (mg s.a./kg) ;
TER = CSEO (mg s.a./kg)/CEPs à long terme (mg s.a./kg).
10.6.1.1. Essais de toxicité aiguë.
Objet de l'essai.
L'essai devrait permettre d'établir la CL 50 et, dans la mesure du possible, la concentration la plus élevée ne provoquant par de mortalité et la concentration la plus faible provoquant 100 p. 100 de mortalité. Les effets morphologiques et de comportement observés doivent être rapportés.
Circonstances où l'essai est requis.
Les essais ne sont requis que lorsque :
- le produit contient plus d'une substance active ;
- la toxicité d'une nouvelle formulation ne peut être prédite avec une fiabilité suffisante à partir d'une formulation testée selon les dispositions de l'annexe I, chapitre 8, point 8.4, ou du présent point.
Lignes directrices.
Les essais doivent être réalisés en conformité avec la méthode 207 de l'OCDE.
10.6.1.2. Essais concernant les effets sublétaux.
Objet de l'essai.
L'essai devrait permettre d'établir la CSEO ainsi que les effets sur la croissance, la reproduction et le comportement.
Circonstances où l'essai est requis.
Les essais ne sont requis que lorsque :
- le produit contient plus d'une substance active ;
- la toxicité d'une nouvelle formulation ne peut être prédite avec une fiabilité suffisante à partir de la formulation testée conformément aux dispositions de l'annexe I, chapitre 8, point 8.4 ;
- la dose recommandée d'application est relevée au-delà de la dose testée précédemment.
Conditions de l'essai.
Les dispositions prévues aux paragraphes correspondants de l'annexe I, chapitre 8, point 8.4.2 sont applicables.
10.6.1.3. Essais de terrain.
Objet de l'essai.
L'essai devrait permettre d'obtenir des données suffisantes pour évaluer les effets sur les vers de terre dans les conditions réelles d'utilisation.
Circonstances où l'essai est requis.
Lorsque TER < 5, il y a lieu d'effectuer un essai de terrain pour déterminer les effets dans les conditions pratiques d'utilisation et d'en rapporter les résultats. Un jugement d'expert est requis pour décider de la nécessité d'effectuer une étude sur la teneur en résidus des vers de terre.
Conditions de l'essai.
Les champs retenus doivent comporter une population de vers de terre raisonnable. L'essai doit être réalisé dans les conditions d'utilisation proposées à la dose maximale proposée. Un produit toxique de référence doit être inclus dans l'essai.
10.6.2. Effets sur d'autres macro-organismes non cibles du sol.
Objet de l'essai.
L'essai devrait permettre d'obtenir des données suffisantes pour évaluer l'impact du produit phytopharmaceutique sur les macro-organismes qui contribuent à la décomposition des plantes mortes et de la matière organique d'origine animale.
Circonstances où l'essai est requis.
L'essai n'est pas requis lorsque, conformément à l'annexe II, chapitre 9, point 9.1, il est démontré que les valeurs de TD 90 sont inférieures à 100 jours, ou que la nature et le mode d'utilisation du produit phytopharmaceutique ne provoquent pas d'exposition ou que les données fournies par les études de la substance active effectuées conformément aux dispositions de l'annexe I, chapitre 8, points 8.3.2, 8.4 et 8.5, indiquent une absence de risques pour la macrofaune, les lombrics ou la microflore du sol.
Lorsque les valeurs de TD 70 f déterminées par les études de dissipation dans le sol (chapitre 9, point 9.1) sont > 365 jours, il y a lieu de déterminer l'impact sur la décomposition de la matière organique.
10.7. Effets sur les micro-organismes non cibles du sol.
10.7.1. Essai en laboratoire.
Objet de l'essai.
L'essai devrait permettre d'obtenir des données suffisantes pour évaluer l'impact du produit phytopharmaceutique sur l'activité microbienne du sol exprimée par la transformation de l'azote et la minéralisation du carbone.
Circonstances où l'essai est requis.
Lorsque les valeurs de TD 90 f déterminées par les études de dissipation dans le sol (chapitre 9, point 9.1) sont > 100 jours, il y a lieu de déterminer l'impact sur les micro-organismes non cibles du sol par des essais de laboratoire. Les essais ne sont cependant pas requis si les études réalisées en conformité avec les dispositions de l'annexe I, chapitre 8, point 8.5, ont fait apparaître des déviations qui, après 100 jours, sont inférieures à 25 p. 100 par rapport aux valeurs témoins de l'activité métabolique de la biomasse microbienne, et si ces données sont compatibles avec les utilisations, la nature et les propriétés de la préparation particulière à autoriser.
Lignes directrices.
Setac. Procédures d'évaluation de l'écotoxicité et du sort des pesticides dans l'environnement.
10.7.2. Essais supplémentaires.
Objet de l'essai.
L'essai devrait permettre d'obtenir des données suffisantes pour évaluer l'impact du produit phytopharmaceutique sur l'activité microbienne dans les conditions d'utilisation pratique.
Circonstances où l'essai est requis.
Lorsque, après 100 jours, l'activité mesurée s'écarte de plus de 25 p. 100 du témoin dans le cadre de l'essai en laboratoire, des essais supplémentaires en laboratoire, en serre et/ou sur le terrain peuvent se révéler nécessaires.
10.8. Données provenant des dépistages biologiques primaires sous forme succincte.
Il y a lieu d'établir un résumé des données produites par les essais préliminaires pratiqués afin d'évaluer l'activité biologique et les doses exploratoires, qu'elles soient positives ou négatives ; ce résumé doit fournir des informations sur l'impact éventuel sur les espèces non cibles (flore et faune) et doit être accompagné d'un avis critique sur la pertinence d'un impact potentiel sur les espèces non cibles.
Article Annexe II
Modifié par Arrêté 1996-02-01 art. 2 JORF 20 février 1996
Modifié par Arrêté 1996-09-24 art. 6 JORF 27 octobre 1996
11. Résumé et évaluation des sections 9 et 10.
Il y a lieu d'établir un résumé et une évaluation de toutes les données visées dans les chapitres 9 et 10, dont le format soit conforme aux instructions données par les autorités compétentes des Etats membres. Ils doivent être accompagnés d'une évaluation détaillée et critique desdites données qui réponde aux orientations et critères d'appréciation et de décision en mettant l'accent, en particulier, sur les risques et dangers réels et potentiels présentés pour l'environnement et pour les espèces non cibles, et apprécie l'ampleur, la qualité et la fiabilité de la base de données. Une attention particulière doit être accordée aux points suivants :
- la prévision de la distribution et du sort dans l'environnement et l'indication des durées correspondantes ;
- l'identification des espèces et des populations non cibles exposées à un risque et la prévision de l'ampleur de l'exposition potentielle ;
- l'évaluation des risques à court et à long terme pour les espèces non cibles - populations, communautés, processus - selon le cas ;
- l'évaluation des risques de destruction de poissons ou de mortalité chez les grands vertébrés ou les prédateurs terrestres, indépendamment des effets à l'échelle de la population ou de la communauté ;
- la définition des précautions nécessaires pour éviter ou réduire au minimum la contamination de l'environnement et pour assurer la protection des espèces non cibles.
Article Annexe II En savoir plus sur cet article...
Modifié par Arrêté 1996-02-01 art. 2 JORF 20 février 1996
12.1. Informations sur les autorisations accordées dans d'autres pays.
12.2. Information sur les limites maximales de résidus (L.M.R.) existant dans d'autres pays.
12.3. Propositions comportant une justification de la classification et de l'étiquetage proposés conformément aux directives n° 67/548/ C.E.E. et n° 78/631/C.E.E. :
- symbole(s) des dangers ;
- indications relatives aux dangers ;
- phrases types relatives à la nature des risques ;
- phrases types relatives aux conseils de prudence.
12.4. Propositions concernant les phrases types relatives à la nature des risques et aux conseils de prudence conformément à l'article 34, points f et g, et étiquetage proposé.
12.5. Spécimens de l'emballage proposé.
Article Annexe II En savoir plus sur cet article...
Modifié par Arrêté 1996-02-01 art. 2 JORF 20 février 1996
3. L'information demandée doit comprendre la classification et l'étiquetage proposés du produit phytosanitaire conformément aux directives communautaires pertinentes.
Partie B : Préparations de micro-organismes ou de virus
Introduction.
i) La présente partie énonce les données requises pour l'autorisation des produits phytopharmaceutiques à base de préparations de micro-organismes, y compris de virus.
La définition du terme "micro-organisme" telle qu'elle figure dans l'introduction de l'annexe I, partie B, s'applique également à l'annexe II, partie B.
ii) Le cas échéant, les données sont analysées à l'aide de méthodes statistiques appropriées. Les analyses statistiques doivent être rapportées de manière circonstanciée (par exemple, toutes les estimations ponctuelles doivent être délimitées par un intervalle de confiance et il y a lieu de fournir les valeurs de probabilité exactes plutôt que d'utiliser la mention "significatif/non significatif".
iii) Dans l'attente de l'adoption de procédures spécifiques au niveau international, les informations requises seront obtenues en appliquant les procédures de test adoptées par l'autorité compétente (comme celles de l'USEPA, par exemple) ; le cas échéant, il y a lieu d'adapter les procédures décrites à l'annexe I, partie A, pour qu'elles puissent convenir aux micro-organismes. Les tests doivent porter sur des micro-organismes viables et, le cas échéant, non viables et comporter un contrôle à blanc.
iv) Si une étude comporte l'utilisation de doses différentes, la relation entre la dose et l'effet néfaste doit être notée.
v) Pour les tests effectués, il y a lieu de fournir une description détaillée (spécifications techniques) du matériel utilisé et des impuretés qu'il contient, conformément aux dispositions de la section 1, point 1.4.
vi) Lorsqu'une nouvelle préparation doit être examinée, l'extrapolation à partir de l'annexe I, partie B, est acceptable, à condition que tous les effets possibles des adjuvants et autres composants, notamment sur le pouvoir pathogène et infectieux, soient également évalués.
Article Annexe II En savoir plus sur cet article...
Créé par Arrêté 2002-12-17 art. 2 JORF 14 janvier 2003
1. Identité du produit phytopharmaceutique.
Les informations fournies, conjuguées aux données requises pour les micro-organismes, doivent être suffisantes pour permettre une identification et une définition précises des préparations. Les informations et données mentionnées, sauf spécification contraire, sont nécessaires pour tous les produits phytopharmaceutiques. L'objectif est en effet de déterminer si un facteur quelconque peut modifier les propriétés du micro-organisme en tant que produit phytopharmaceutique, par opposition au micro-organisme en tant que tel, qui fait l'objet de l'annexe 1, partie B, du présent arrêté.
1.1. Demandeur.
Le nom et l'adresse du demandeur (adresse permanente dans la Communauté) doivent être indiqués, tout comme les nom, qualité, numéros de téléphone et de télécopieur de la personne à contacter.
Si, en outre, le demandeur dispose d'un bureau, d'un agent ou d'un représentant en France, le nom et l'adresse du bureau, de l'agent ou du représentant local doivent être fournis ainsi que les nom, qualité, numéros de téléphone et de télécopieur de la personne à contacter.
1.2. Fabricant de la préparation et du ou des micro-organismes.
Le nom et l'adresse du fabricant de la préparation et de chaque micro-organisme contenu dans celle-ci doivent être fournis, tout comme le nom et l'adresse de chaque installation dans laquelle la préparation et le micro-organisme sont produits.
Un point de contact (de préférence un point de contact central avec nom, numéros de téléphone et de télécopieur) doit être indiqué pour chaque fabricant.
Lorsque le micro-organisme provient d'un fabricant pour lequel les données prévues à l'annexe I, partie B, n'ont pas été communiquées, les informations détaillées requises à l'annexe I, partie B, section 1.3, en ce qui concerne le nom et la description des espèces ainsi que celles requises à l'annexe I, partie B, section 1.4, en ce qui concerne les impuretés doivent être fournies.
1.3. Nom commercial ou nom commercial proposé et, le cas échéant, numéro de code de développement attribué au fabricant pour la préparation.
Tous les noms commerciaux, anciens et courants, noms commerciaux proposés et numéros de code de développement de la préparation visée dans le dossier ainsi que les noms et numéros courants doivent être fournis. Le détail complet de toute différence éventuelle doit être fourni. (Le nom commercial proposé ne doit pas prêter à confusion avec le nom commercial des produits phytopharmaceutiques déjà autorisés).
1.4. Informations quantitatives et qualitatives détaillées sur la composition de la préparation.
i) Chacun des micro-organismes visés par la demande doit être identifié et désigné par son nom d'espèce. Le micro-organisme doit être déposé auprès d'une banque de collection de cultures de réputation internationale et se voir attribuer un numéro de dépôt. Le nom scientifique doit être indiqué ainsi que l'affectation de groupe (bactéries, virus, etc.) et toute autre dénomination concernant le micro-organisme (par exemple, la souche, le sérotype). En outre, le stade de développement du micro-organisme dans le produit commercialisé (spores ou mycélium, par exemple) doit être précisé.
ii) En ce qui concerne les préparations, les informations suivantes doivent être communiquées :
- la teneur en micro-organisme(s) du produit phytopharmaceutique et la teneur en micro-organisme du matériel utilisé pour la fabrication des produits phytopharmaceutiques. La teneur maximale, la teneur minimale et la teneur nominale du matériel viable et du matériel non viable doivent être précisées ;
- la teneur en adjuvants ;
- la teneur en autres composants (tels que les sous-produits, les condensats, le milieu de culture, etc.) et micro-organismes contaminants, issus du processus de production.
La teneur doit être exprimée selon les termes prévus à l'article 34 (c) du présent arrêté pour les substances chimiques et selon les termes qui conviennent pour les micro-organismes (nombre d'unités actives par unité de volume ou de poids ou toute autre manière adaptée pour le micro-organisme considéré).
iii) Les adjuvants sont, si possible, identifiés soit par le nom chimique indiqué à l'annexe I de l'arrêté du 20 avril 1994 modifié relatif à la déclaration, la classification, l'emballage et l'étiquetage des substances, soit, s'ils n'y figurent pas, conformément aux nomenclatures de l'UICPA et des CA. Leur structure ou formule développée doit être précisée. Pour chaque composant des adjuvants, il faut indiquer, s'ils existent, le numéro CEE (Einecs ou Elincs) et le numéro CAS. Si l'information fournie ne permet par d'identifier parfaitement un adjuvant, des spécifications appropriées doivent être fournies. S'il existe, il y a lieu d'indiquer également le nom commercial des adjuvants.
iv) La fonction des adjuvants doit être indiquée :
- adhésif ;
- agent antimousse ;
- antigel ;
- liant ;
- tampon ;
- agent porteur ;
- désodorisant ;
- agent dispersant ;
- colorant ;
- émétique ;
- émulsifiant ;
- engrais ;
- agent odorant ;
- parfum ;
- conservateur ;
- propulseur ;
- répulsif ;
- phytoprotecteur ;
- solvant ;
- stabilisant ;
- synergiste ;
- épaississant ;
- agent mouillant ;
- divers (à préciser).
iv) Identification des micro-organismes contaminants et d'autres composants issus du processus de production.
Les micro-organismes contaminants doivent être identifiés conformément aux dispositions de l'annexe I, partie B, section 1, point 1.3.
Les substances chimiques (composants inertes, sous-produits, etc.) doivent être identifiées conformément aux dispositions de l'annexe I, partie A, section 1, point 1.10.
Lorsque les informations fournies ne permettent pas d'identifier précisément un composant, tel que le condensat ou le milieu de culture, des informations détaillées doivent être données sur la composition de chacun de ces composants.
1.5. Nature et état physiques de la préparation.
Le type et le code de la préparation doivent être spécifiés conformément au Catalogue des types de formulation de pesticides et système de code international (Monographie technique GIFAP n° 2, 1989).
Lorsqu'une préparation donnée n'est pas définie précisément dans cette publication, il y a lieu de fournir une description complète de la nature et de l'état physiques de la préparation ainsi qu'une proposition de description convenable du type de préparation et une proposition de définition correspondante.
1.6. Fonction.
La fonction biologique doit être retenue parmi les suivantes :
- bactéricide ;
- fongicide ;
- insecticide ;
- acaricide ;
- molluscicide ;
- nématocide ;
- herbicide ;
- autres (à préciser).
2. Propriétés physiques, chimiques et techniques du produit phytopharmaceutique.
Il y a lieu d'indiquer dans quelle mesure les produits phytopharmaceutiques pour lesquels l'autorisation est demandée sont conformes aux spécifications FAO correspondantes, établies par le Groupe d'experts des spécifications relatives aux pesticides du groupe d'experts FAO des spécifications, critères d'homologation et normes d'application des pesticides. Toute différence par rapport aux spécifications FAO doit être décrite en détail et justifiée.
2.1. Aspect (couleur et odeur).
Une description de la couleur et de l'odeur, le cas échéant, ainsi que de l'état physique de la préparation doit être fournie.
2.2. Stabilité de stockage et durée de conservation.
2.2.1. Incidence de la lumière, de la température et de l'humidité sur les caractéristiques techniques du produit phytopharmaceutique.
i) La stabilité physique et biologique de la préparation à la température de stockage recommandée ainsi que la croissance des micro-organismes contaminants doivent être déterminées et décrites. Les conditions de réalisation du test doivent être justifiées.
ii) En outre, pour les préparations liquides, il y a lieu de déterminer et de décrire l'incidence des températures basses sur la stabilité conformément aux méthodes CIMAP MT 39, MT 48, MT 51 ou MT 54, selon le cas.
iii) La durée de conservation de la préparation à la température de stockage recommandée doit être précisée.
Si elle est inférieure à deux ans, il y a lieu d'indiquer cette durée en mois, en donnant les spécifications de température appropriées. La monographie n° 17 du GIFAP contient des informations utiles.
2.2.2. Autres facteurs agissant sur la stabilité.
L'incidence de l'exposition à l'air, à l'emballage, etc., sur la stabilité du produit doit être évaluée.
2.3. Propriétés explosives et oxydantes.
Les propriétés explosives et oxydantes seront déterminées ainsi qu'il est prévu à l'annexe II, partie A, section 2, point 2.2, à moins que l'inutilité d'une telle étude sur le plan technique ou scientifique puisse être démontrée.
2.4. Point d'inflammation et autres indications sur l'inflammabilité ou l'ignition spontanée.
Les propriétés explosives et oxydantes seront déterminées ainsi qu'il est prévu à l'annexe II, partie A, section 2, point 2.3, à moins que l'inutilité d'une telle étude sur le plan technique ou scientifique ne puisse être démontrée.
2.5. Acidité, alcalinité et, si nécessaire, valeur du pH.
L'acidité, l'alcalinité et le pH seront déterminés ainsi qu'il est prévu à l'annexe II, partie A, section 2, point 2.4, à moins que l'inutilité d'une telle étude sur le plan technique ou scientifique ne puisse être démontrée.
2.6. Viscosité et tension superficielle.
La viscosité et la tension superficielle seront déterminées ainsi qu'il est prévu à l'annexe II, partie A, section 2, point 2.5, à moins que l'inutilité d'une telle étude sur le plan technique ou scientifique ne puisse être démontrée.
2.7. Caractéristiques techniques du produit phytopharmaceutique.
Les caractéristiques techniques de la préparation doivent être déterminées en vue d'une décision concernant son acceptabilité. Si des tests sont nécessaires, ils doivent être réalisés à des températures permettant la survie du micro-organisme.
2.7.1. Mouillabilité.
La mouillabilité des préparations solides utilisées en dilution (poudres mouillables et granulés hydrodispersibles, par exemple) doit être déterminée et décrite conformément à la méthode CIMAP MT 53.3.
2.7.2. Formation de mousse persistante.
La persistance de mousse dans les préparations destinées à être diluées dans l'eau doit être déterminée et décrite conformément à la méthode CIMAP MT 47.
2.7.3. Faculté de passer en suspension et stabilité de la suspension.
Il y a lieu de déterminer et de décrire la faculté de passer en suspension des produits hydrodispersibles (poudres mouillables, granulés hydrodispersibles, suspensions concentrées, par exemple) conformément à la méthode CIMAP MT 15, MT 161 ou MT 168, selon le cas.
Pour les produits hydrodispersibles (suspensions concentrées et granulés hydrodispersibles, par exemple), la spontanéité de la dispersion doit être déterminée et décrite conformément aux méthodes CIMAP MT 160 ou MT 174, selon le cas.
2.7.4. Test du tamis humide, test du tamis sec.
Afin de garantir une distribution granulométrique des particules dans les poudres pour poudrage qui rende leur utilisation aisée, il y a lieu d'effectuer un test du tamis sec et de le décrire conformément à la méthode CIMAP MT 59.1.
S'il s'agit de produits hydrodispersibles, un test du tamis humide doit être réalisé et décrit conformément à la méthode CIMAP MT 59.3 ou MT 167, selon le cas.
2.7.5. Distribution granulométrique (poudres pour poudrage et mouillables, granulés), teneur en poussières/particules fines (granulés), usure et friabilité (granulés).
i) S'il s'agit de poudres, la distribution granulométrique des particules doit être déterminée et décrite conformément à la méthode 110 de l'OCDE.
La granulométrie nominale des granulés destinés à une application directe doit être déterminée et décrite conformément à la méthode CIMAP MT 58.3 et celle des granulés hydrodispersibles conformément à la méthode CIMAP MT 170.
ii) La teneur en poussières des préparations granulées doit être déterminée et décrite conformément à la méthode CIMAP MT 171. S'il convient d'évaluer l'exposition de l'opérateur, la taille des particules de poussière doit être déterminée et décrite conformément à la méthode 110 de l'OCDE.
iii) Les caractéristiques de friabilité et d'usure des granulés seront déterminées et décrites dès que des méthodes auront été adoptées au niveau international. Si des données sont déjà disponibles, elles doivent être indiquées, ainsi que la méthode utilisée.
2.7.6. Emulsibilité, réémulsibilité et stabilité de l'émulsion.
i) L'émulsibilité, la stabilité de l'émulsion et la réémulsibilité des préparations sous forme d'émulsions doivent être déterminées et décrites conformément aux méthodes CIMAP MT 36 ou MT 173, selon le cas.
ii) La stabilité des émulsions diluées et des préparations sous forme d'émulsions doit être déterminée et décrite conformément à la méthode CIMAP MT 20 ou MT 173.
2.7.7. Faculté d'écoulement, de déversement (faculté de rinçage) et de transformation en poussières.
i) La faculté d'écoulement des préparations granulées doit être déterminée et décrite conformément à la méthode CIMAP MT 172.
ii) La faculté de déversement (résidu de rinçage y compris) des suspensions (suspensions concentrées ou suspoémulsions, par exemple) doit être déterminée et décrite conformément à la méthode CIMAP MT 148.
iii) La faculté de transformation en poussières doit être déterminée et décrite conformément à la méthode CIMAP MT 34 ou à toute autre méthode appropriée.
2.8. Compatibilité physique, chimique et biologique avec d'autres produits, y compris les produits phytopharmaceutiques, avec lesquels il est prévu d'autoriser l'utilisation de la préparation.
2.8.1. Compatibilité physique.
La compatibilité physique des mélanges en cuve recommandés doit être déterminée et décrite.
2.8.2. Compatibilité chimique.
La compatibilité chimique des mélanges en cuve recommandés doit être déterminée et décrite, sauf lorsque l'examen des propriétés particulières des préparations établit avec un degré de certitude suffisant qu'aucune réaction ne peut avoir lieu. Dans ce cas, il suffit de donner cette information pour justifier l'inutilité d'une détermination effective de la compatibilité chimique.
2.8.3. Compatibilité biologique.
La compatibilité biologique des mélanges en cuve doit être déterminée et décrite. Les effets (antagonisme ou effets fongicides, par exemple) sur l'activité du micro-organisme après mélange avec d'autres micro-organismes ou substances chimiques doivent être décrits. Il convient d'étudier, sur la base des données relatives à l'efficacité, l'interaction possible du produit phytopharmaceutique avec d'autres produits chimiques à appliquer sur les cultures dans les conditions prévues d'utilisation de la préparation. Afin d'éviter toute perte d'efficacité, il y a lieu de spécifier, le cas échéant, les intervalles à respecter entre l'application du pesticide biologique et celle des pesticides chimiques.
2.9. Adhérence et répartition sur semences.
Lorsque les préparations sont destinées au traitement des semences, tant la répartition que l'adhérence doivent être étudiées et décrites ; dans le cas de la répartition, il faut procéder conformément à la méthode CIMAP MT 175.
2.10. Résumé et évaluation des données fournies au titre des points 2.1 à 2.9.
3. Données relatives à l'application.
3.1. Usage agricole envisagé.
Préciser le ou les usages agricoles actuels et proposés des préparations contenant le micro-organisme, parmi les usages du catalogue des usages agricoles.
3.2. Mode d'action.
Les voies possibles d'absorption du produit (contact, ingestion ou inhalation, par exemple) ou l'action antiparasitaire (action fongitoxique, action fongistatique, compétition nutritionnelle, etc.) doivent être précisées.
Il y a lieu d'indiquer également si le produit subit une translocation dans les végétaux et, le cas échéant, si cette translocation est apoplastique, symplastique ou les deux.
3.3. Modalités de l'utilisation prévue.
Les modalités de l'utilisation prévue, par exemple les types d'organismes nuisibles à combattre et/ou les végétaux ou les produits végétaux à protéger, doivent être spécifiées.
Il convient également d'indiquer les intervalles à respecter entre l'application du produit phytopharmaceutique contenant des micro-organismes et celle des pesticides chimiques, ou bien de fournir une liste des substances actives présentes dans les produits phytopharmaceutiques chimiques à ne pas utiliser avec le produit phytopharmaceutique contenant des micro-organismes sur la même culture.
3.4. Dose d'application.
Pour chaque méthode d'application et chaque utilisation, la dose d'application par unité traitée (ha, m2, m3) doit être spécifiée en grammes, kilogrammes ou litres pour la préparation et dans des unités appropriées pour le micro-organisme.
Les doses d'application sont normalement exprimées en grammes ou kilogrammes/hectare ou encore en kilogrammes/mètre cube et, le cas échéant, en grammes ou kilogrammes/tonne ; pour les serres et les jardins domestiques, les doses d'utilisation sont indiquées en grammes ou kilogrammes/100 mètres carrés ou en grammes ou kilogrammes/mètres cubes.
3.5. Teneur en micro-organismes du matériel utilisé (par exemple dans le produit de pulvérisation dilué, les appâts ou les semences traitées).
La teneur en micro-organismes est spécifiée en nombre d'unités actives/millilitre, en grammes ou dans toute autre unité appropriée, selon le cas.
3.6. Méthode d'application.
Il y a lieu de décrire in extenso la méthode d'application, en indiquant, le cas échéant, le type d'équipement à utiliser ainsi que le type et le volume de diluant à utiliser par unité de surface ou de volume.
3.7. Nombre et calendrier des applications et durée de la protection.
Il convient d'indiquer le nombre maximal d'applications avec leur calendrier. Le cas échéant, les stades de développement de la culture ou des végétaux à protéger ainsi que ceux des organismes nuisibles doivent également être spécifiés. Si possible et si nécessaire, il y a lieu de préciser en nombre de jours l'intervalle à respecter entre deux applications.
Indiquer également la durée de protection assurée pour chaque application et pour le nombre maximal d'applications.
3.8. Périodes d'attente nécessaires ou autres précautions à prendre pour éviter tout effet phytopathogène sur les cultures ultérieures.
Le cas échéant, il convient d'indiquer, sur la base des données prévues à la section 6, point 6.6, la période d'attente minimale nécessaire entre la dernière application et l'ensemencement ou la plantation des cultures suivantes pour prévenir tout effet phytopathogène sur ces dernières.
Indiquer les limitations éventuelles quant au choix des cultures suivantes.
3.9. Consignes d'utilisation proposées.
Les consignes d'utilisation proposées de la préparation, à imprimer sur des étiquettes et des notices, doivent être spécifiées.
Article Annexe II
Créé par Arrêté 2002-12-17 art. 2 JORF 14 janvier 2003
4.1. Emballage et compatibilité de la préparation avec les matériaux d'emballage proposés.
i) L'emballage à utiliser doit être décrit et spécifié de manière exhaustive, en précisant les matériaux utilisés, le mode de fabrication (par exemple, extrusion, soudage, etc.), la taille et la capacité, la taille de l'ouverture, le type de fermeture et le scellement. Il doit être conçu conformément aux critères et aux lignes directrices spécifiés dans les directives pour le conditionnement et le stockage des pesticides de la FAO.
ii) L'adéquation de l'emballage, y compris les dispositifs de fermeture, sur le plan de la solidité, de l'imperméabilité et de la résistance à des conditions de transport et de manutention normale, doit être déterminée et décrite conformément aux méthodes ADR 3552, 3553, 3560, 3554, 3555, 3556, 3558 ou aux méthodes ADR appropriées pour les grands récipients à vrac et, si des fermetures de sécurité pour les enfants et les dispositifs permettant de détecter les dangers au toucher sont nécessaires pour la préparation considérée, conformément à l'annexe IX de l'arrêté du 20 avril 1994 modifié relatif à la déclaration, la classification, l'emballage et l'étiquetage des substances (concerne les dispositions relatives aux fermetures de sécurité pour les enfants et la détection des dangers au toucher).
iii) La résistance du matériau d'emballage à son contenu doit être spécifiée ainsi qu'indiqué dans la monographie GIFAP n° 17.
4.2. Procédures de nettoyage de l'équipement utilisé pour les applications.
Les procédures à mettre en œuvre pour le nettoyage de l'équipement d'application et des vêtements de protection doivent être décrites en détail. L'efficacité de la procédure de nettoyage doit être déterminée, à l'aide de tests biologiques par exemple, puis rapportée.
4.3. Délais de retour, délais d'attente nécessaires ou autres précautions à prendre pour protéger les personnes, le bétail et l'environnement.
Les informations communiquées doivent découler et être corroborées par les données fournies pour le ou les micro-organismes et celles visées aux sections 7 et 8.
i) Le cas échéant, il y a lieu de spécifier les délais d'attente avant récolte, les délais de retour et les délais de retrait nécessaires pour réduire au maximum la présence de résidus dans ou sur les récoltes, végétaux et produits végétaux ou dans des espaces ou emplacements traités, en vue de protéger les personnes et le bétail. Il s'agit par exemple :
- du délai d'attente avant récolte (en jours) pour chaque culture concernée ;
- du délai de retour (en jours) du bétail dans les zones de pâturage ;
- du délai de retour (en heures ou en jours) de l'homme dans les cultures, les bâtiments ou les espaces traités ;
- du délai de retrait (en jours) des aliments pour animaux ;
- du délai d'attente (en jours) entre l'application et la manipulation des produits traités.
ii) Si nécessaire, compte tenu des résultats des essais, il convient de fournir des informations sur les conditions agronomiques, phytosanitaires ou environnementales particulières dans lesquelles la préparation peut ou ne peut pas être utilisée.
4.4. Méthodes et précautions recommandées en matière de manipulation, de stockage, de transport ou en cas d'incendie.
Les méthodes et les précautions recommandées en ce qui concerne les procédures de manipulation (détaillées) en vue du stockage, aussi bien au niveau de l'entrepôt que de l'utilisateur, des produits phytopharmaceutiques en vue de leur transport et en cas d'incendie doivent être indiquées. Il y a lieu, le cas échéant, de fournir des informations relatives aux produits de combustion. Spécifier les risques probables ainsi que les méthodes et les procédures à mettre en oeuvre en vue de minimiser les dangers. Il y a également lieu d'indiquer les procédures à observer en vue de prévenir ou de minimiser la formation de déchets ou tout phénomène de rémanence. Le cas échéant, il convient de procéder à une évaluation conformément à la norme IS0 TR 9122.
La nature et les caractéristiques des vêtements de protection et de l'équipement proposés doivent être précisées. Les informations fournies doivent permettre d'évaluer leur adéquation et leur efficacité dans des conditions d'utilisation réalistes (par exemple, dans les champs ou sous serres).
4.5. Mesures en cas d'accident.
Les modalités des mesures à mettre en oeuvre en cas d'accident au cours du transport, du stockage ou de l'utilisation doivent être précisées et comprennent :
- la contention des déversements ;
- la décontamination des terres, des véhicules et des bâtiments ;
- l'élimination des emballages endommagés, des absorbants et autres matériaux ;
- la protection du personnel d'intervention et des assistants ;
- les mesures de premiers secours.
4.6. Procédures de destruction ou de décontamination du produit phytopharmaceutique et de son emballage.
Les procédures de destruction et de décontamination doivent être mises au point pour les petites quantités (niveau de l'utilisateur) et les grandes quantités (niveau de l'entrepôt). Les procédures doivent être conformes aux dispositions en vigueur concernant l'élimination des déchets et notamment des déchets toxiques. Les moyens d'élimination proposés ne doivent pas avoir d'incidence inacceptable sur l'environnement et doivent constituer les moyens d'élimination les plus pratiques et les plus efficaces possibles sur le plan des coûts.
4.6.1. Incinération contrôlée.
Dans de nombreux cas, le meilleur ou l'unique moyen d'éliminer en toute sécurité les produits phytopharmaceutiques, et notamment les adjuvants qu'ils contiennent, les matériaux contaminés ou les emballages contaminés, est de les soumettre à une incinération contrôlée dans un incinérateur agréé. Le demandeur est tenu de fournir les consignes nécessaires pour garantir la sécurité de l'opération.
4.6.2. Autres méthodes.
Décrire de manière exhaustive les autres méthodes d'élimination des produits phytopharmaceutiques, des emballages et des matériaux contaminés, lorsqu'elles sont proposées. Fournir des informations concernant ces méthodes en vue d'établir leur efficacité et leur sûreté.
5. Méthodes d'analyse.
Introduction.
Les dispositions de la présente section s'appliquent exclusivement aux méthodes d'analyse requises pour le contrôle et le suivi postérieurs à l'autorisation.
Dans toute la mesure du possible, il est souhaitable que les produits phytopharmaceutiques soient exempts de contaminants. Le niveau des contaminants acceptables doit être établi par l'autorité compétente sur la base d'une évaluation des risques.
Le demandeur doit assurer un contrôle de qualité continu tant du processus de production que du produit obtenu. Il convient de soumettre les critères de qualité applicables au produit.
En ce qui concerne les méthodes d'analyse utilisées pour la production des données requises par le présent arrêté ou à d'autres fins, le demandeur est tenu de fournir une justification de la méthode utilisée. Si nécessaire, des arrêtés spécifiques seront élaborés pour ces méthodes sur la base des mêmes normes que celles requises pour les méthodes de contrôle et de suivi postérieurs à l'autorisation.
Il convient de fournir une description des méthodes d'analyse contenant toutes les données utiles relatives à l'équipement et au matériel utilisés ainsi qu'aux conditions d'application. L'applicabilité des méthodes CIMAP actuelles doit être rapportée. Ces méthodes doivent, autant que possible, suivre l'approche la plus simple, être peu onéreuses et faire appel à des équipements courants.
Les définitions mentionnées ci-après s'appliquent aux fins de la présente section.
Impuretés : tout composant (y compris les micro-organismes contaminants et/ou les substances chimiques) autres que le micro-organisme désigné, provenant du processus de fabrication ou d'une dégradation intervenue en cours de stockage.
Impuretés sensibles : impuretés, telles que définies ci-dessus, qui présentent un risque pour la santé humaine ou animale et/ou pour l'environnement.
Métabolites : produits qui résultent des réactions de dégradation et des réactions biosynthétiques intervenant dans le micro-organisme ou dans tout autre organisme utilisé pour la production du micro-organisme concerné.
Métabolites sensibles : métabolites qui présentent un risque pour la santé humaine ou animale et/ou pour l'environnement.
Résidus : micro-organismes viables et substances fabriquées en quantité significative par les micro-organismes, qui persistent après la disparition des micro-organismes et présentent un risque pour la santé humaine ou animale et/ou l'environnement.
Les échantillons suivants doivent être fournis sur demande :
i) Echantillons de la préparation ;
ii) Echantillons du micro-organisme tel qu'il est produit ;
iii) Etalons pour l'analyse du micro-organisme pur ;
iv) Etalons pour l'analyse des métabolites sensibles et de tous les autres composants compris dans la définition de résidu ;
v) S'ils sont disponibles, des échantillons des substances de référence des impuretés sensibles.
5.1. Méthodes d'analyse de la préparation.
Il y a lieu de fournir et de décrire de manière exhaustive les méthodes d'identification et de détermination de la teneur en micro-organisme de la préparation. Dans le cas d'une préparation contenant plusieurs micro-organismes, il convient d'indiquer les méthodes permettant d'identifier et de déterminer la teneur de chacun d'entre eux.
Méthodes permettant d'assurer un contrôle régulier du produit final (préparation) afin de veiller à ce qu'il ne contienne pas d'autres organismes que ceux indiqués et de garantir son uniformité.
Méthodes d'identification des micro-organismes contaminants de la préparation.
Il y a lieu de préciser les méthodes employées pour déterminer la stabilité de stockage et la durée de conservation de la préparation.
5.2. Méthodes de détermination et de quantification des résidus.
Il y a lieu de présenter des méthodes d'analyse pour la détermination des résidus, conformément à l'annexe I, partie B, section 4, point 4.2, sauf s'il est établi que les informations déjà soumises en vertu de l'annexe I, partie B, section 4, point 4.2, sont suffisantes.
6. Données d'efficacité.
Les données relatives à l'efficacité à fournir sont définies dans la partie A de cette annexe, section 6.
7. Effets sur la santé humaine.
Afin que la toxicité, notamment la pathogénicité et l'infectiosité des préparations, puisse être dûment évaluée, il convient de disposer d'informations suffisantes en ce qui concerne la toxicité aiguë du micro-organisme, ainsi que les phénomènes d'irritation et de sensibilisation dont il peut être responsable. Dans la mesure du possible, des informations supplémentaires sur le mode d'action toxique, le profil toxicologique et tout autre aspect toxicologique connu du micro-organisme doivent être présentées. Il y a lieu d'accorder une attention particulière aux adjuvants.
Les études toxicologiques doivent faire état de tout signe d'infection ou de pathogénicité. Elles doivent également explorer les moyens d'élimination.
Compte tenu de l'influence que les impuretés et d'autres composants peuvent exercer sur le comportement toxicologique, il est essentiel de fournir, pour toute étude proposée, une description détaillée (spécifications techniques) du matériel utilisé. Des tests doivent être effectués avec le produit phytopharmaceutique à autoriser. En particulier, il doit être clair que le micro-organisme utilisé dans la préparation et les conditions dans lesquelles il est cultivé sont identiques au micro-organisme et aux conditions de culture pour lesquels les informations et données sont soumises dans le cadre de l'annexe I, partie B.
L'étude du produit phytopharmaceutique sera effectuée sur la base d'essais séquentiels.
7.1. Etudes de base sur la toxicité aiguë.
Les études, les données et les informations à fournir et à évaluer doivent être suffisantes pour permettre d'apprécier les effets d'une exposition unique au produit phytopharmaceutique et, en particulier, pour déterminer ou indiquer :
- la toxicité du produit phytopharmaceutique ;
- la toxicité du produit phytopharmaceutique par rapport au micro-organisme ;
- l'évolution au cours du temps et les caractéristiques des effets, avec description exhaustive des modifications comportementales et des éventuelles constatations macropathologiques à l'inspection post mortem ;
- si possible, le mode d'action toxique, ainsi que ;
- les risques relatifs liés aux diverses voies d'exposition.
Si l'accent doit être placé sur l'estimation des niveaux de toxicité considérés, les informations obtenues doivent aussi permettre la classification du produit phytopharmaceutique conformément à l'arrêté du 20 février 1990 modifié définissant les critères de classification et les conditions d'étiquetage et d'emballage des préparations dangereuses. Les informations obtenues grâce aux tests de toxicité aiguë sont particulièrement utiles pour évaluer les dangers potentiels en cas d'accident.
7.1.1. Toxicité orale aiguë.
Situations dans lesquelles les tests sont requis :
Un test de toxicité orale aiguë doit toujours être effectué.
Ligne directrice pour les tests :
Les tests doivent être effectués conformément à la méthode B 1 ou B 1 bis de l'annexe V de la directive 67/548/CEE relative à la classification, l'emballage et l'étiquetage des substances dangereuses, modifiée notamment par la directive 92/69/CEE.
7.1.2. Toxicité aiguë par inhalation.
Objet du test :
Le test a pour objectif de déterminer la toxicité par inhalation du produit phytopharmaceutique chez les rats.
Situations dans lesquelles les tests sont requis :
Le test doit être effectué lorsque le produit phytopharmaceutique :
- est utilisé à l'aide d'un équipement de nébulisation, est un aérosol ;
- est une poudre contenant une proportion significative de particules d'un diamètre 50 micromètres (1 % sur la base du poids) ;
- est appliqué par voie aérienne dans le cas où une exposition par inhalation est possible ;
- est appliqué selon un procédé induisant l'apparition d'une proportion significative de particules ou de gouttelettes d'un diamètre 50 micromètres (1 % sur la base du poids) ;
- contient un composant volatil à concurrence de plus de 10 %.
Ligne directrice pour les tests :
Les tests doivent être réalisés conformément à la méthode B 2 de l'annexe V de la directive 67/548/CEE relative à la classification, l'emballage et l'étiquetage des substances dangereuses, modifiée notamment par les directives 92/69/CEE et 93/21/CEE.
7.1.3. Toxicité percutanée aiguë.
Situations dans lesquelles les tests sont requis.
Un test de toxicité percutanée aiguë doit toujours être effectué.
Ligne directrice pour les tests :
Les tests doivent être réalisés conformément à la méthode B 3 de l'annexe V de la directive 67/548/CEE relative à la classification, l'emballage et l'étiquetage des substances dangereuses, modifiée notamment par la directive 92/69/CEE.
7.2. Etudes complémentaires sur la toxicité aiguë.
7.2.1. Irritation cutanée.
Objet du test :
Le test a pour objectif d'évaluer le pouvoir irritant pour la peau du produit phytopharmaceutique, y compris la réversibilité potentielle des effets observés.
Situations dans lesquelles les tests sont requis :
Le pouvoir irritant du produit phytopharmaceutique doit toujours être déterminé, sauf lorsque les adjuvants ne sont pas supposés irriter la peau ou lorsqu'il est démontré que le micro-organisme n'irrite pas la peau, ou encore lorsque tout risque grave pour la peau peut être raisonnablement écarté, ainsi qu'indiqué dans les directives d'exécution des tests.
Ligne directrice pour les tests :
Les tests doivent être réalisés conformément à la méthode B 4 de l'annexe V de la directive 67/548/CEE relative à la classification, l'emballage et l'étiquetage des substances dangereuses, modifiée notamment par la directive 92/69/CEE.
7.2.2. Irritation oculaire.
Objet du test :
Le test a pour objectif d'évaluer le pouvoir irritant pour les yeux du produit phytopharmaceutique, y compris la réversibilité potentielle des effets observés.
Situations dans lesquelles les tests sont requis :
Le pouvoir irritant pour les yeux du produit phytopharmaceutique doit être déterminé lorsque les adjuvants sont suspectés d'entraîner une irritation oculaire, sauf dans les cas où le micro-organisme est irritant pour l'œil ou s'il est probable, ainsi qu'indiqué dans les lignes directrices pour les tests, que l'œil subisse des dommages graves.
Ligne directrice pour les tests :
L'irritation oculaire doit être évaluée conformément à la méthode B 5 de l'annexe V de la directive 67/548/CEE relative à la classification, l'emballage et l'étiquetage des substances dangereuses, modifiée notamment par la directive 92/69/CEE.
7.2.3. Sensibilisation cutanée.
Objet du test :
Le test a pour objectif de fournir des informations suffisantes pour évaluer la capacité du produit phytopharmaceutique de provoquer des réactions de sensibilisation cutanée.
Situations dans lesquelles les tests sont requis :
Les tests doivent être effectués lorsque les adjuvants sont suspectés d'avoir des propriétés de sensibilisation cutanée, sauf dans les cas où il est établi que le ou les micro-organismes ou les adjuvants ont des propriétés de sensibilisation cutanée.
Ligne directrice pour les tests :
Les tests doivent être effectués conformément à la méthode B 6 de l'annexe V de la directive 67/548/CEE relative à la classification, l'emballage et l'étiquetage des substances dangereuses, modifiée notamment par la directive 96/54/CE.
7.3. Données relatives à l'exposition.
Les risques pour les personnes en contact avec des produits phytopharmaceutiques (opérateurs, tiers, travailleurs) dépendent des propriétés physiques, chimiques et toxicologiques du produit phytopharmaceutique concerné ainsi que du type de produit (non dilué/dilué), du type de formulation et de la voie, du degré et de la durée d'exposition. Des informations et des données suffisantes doivent être recueillies et rapportées afin de permettre d'évaluer l'importance de l'exposition au produit phytopharmaceutique susceptible de survenir dans les conditions d'utilisation proposées. Lorsqu'il y a lieu de craindre une possible absorption par voie cutanée sur la base des informations fournies sur le micro-organisme à l'annexe I, partie B, section 5, ou d'après les informations concernant la préparation, figurant à l'annexe II, partie B, présente section, des données complémentaires sur l'absorption par voie cutanée peuvent se révéler nécessaires. Les résultats du contrôle de l'exposition pendant la production ou l'utilisation du produit doivent être communiqués. Les informations et les données susmentionnées doivent servir de base à la sélection des mesures de protection appropriées, y compris les équipements de protection individuelle à utiliser par les opérateurs et les travailleurs et à spécifier sur l'étiquette.
7.4. Données toxicologiques disponibles relatives aux substances non actives.
Il y a lieu de présenter, pour chacun des adjuvants, une copie de la notification et de la fiche de données de sécurité visées aux articles R. 231-53 et R. 231-53-1 du code du travail. Toutes les autres informations disponibles doivent également être communiquées.
7.5. Etudes complémentaires relatives aux associations de produits phytopharmaceutiques.
Objet du test :
Il peut parfois se révéler nécessaire d'effectuer les études visées aux points 7.1 à 7.2.3 en cas d'association de plusieurs produits phytopharmaceutiques, lorsque l'étiquette du produit compte des indications d'utilisation du produit phytopharmaceutique avec d'autres produits phytopharmaceutiques et/ou avec des adjuvants mélangés dans le réservoir de l'appareil de pulvérisation. Les décisions concernant la nécessité d'études complémentaires doivent être prises cas par cas, compte tenu des résultats des études de toxicité aiguë relatives aux différents produits phytopharmaceutiques, de la possibilité d'une exposition aux produits associés en cause et des informations disponibles ou de l'expérience pratique concernant les produits en cause ou des produits similaires.
7.6. Synthèse et évaluation des effets sur la santé.
Une synthèse de toutes les données et informations fournies en application des points 7.1 à 7.5 doit être présentée ; elle doit comporter une évaluation détaillée et critique desdites données sur la base de critères et de lignes directrices pertinentes concernant l'évaluation et la prise de décision, compte tenu particulièrement des risques potentiels ou effectifs pour les êtres humains et les animaux ainsi que de l'ampleur, de la qualité et de la fiabilité de la base de données.
8. Résidus dans ou sur les produits traités, les denrées alimentaires et les aliments pour animaux.
Les mêmes dispositions que celles visées à l'annexe I, partie B, section 6, sont applicables ; les informations requises en vertu de la présente section doivent être fournies, à moins qu'il ne soit possible d'extrapoler le comportement du produit phytopharmaceutique sur le plan de la persistance de résidus à partir des données disponibles pour le micro-organisme. Une attention particulière doit être accordée à l'influence des substances comprises dans la préparation sur le comportement du micro-organisme et de ses métabolites, en ce qui concerne la persistance de résidus.
9. Devenir et comportement dans l'environnement.
Les mêmes dispositions que celles visées à l'annexe I, partie B, section 7, sont applicables ; les informations requises en vertu de la présente section doivent être fournies, à moins qu'il ne soit possible d'extrapoler le devenir et le comportement du produit phytopharmaceutique dans l'environnement sur la base des données disponibles à l'annexe I, partie B, section 7.
10. Effets sur les organismes non visés.
Introduction.
i) Les informations fournies, jointes à celles qui concernent le ou les micro-organismes doivent être suffisantes pour permettre d'évaluer les effets du produit phytopharmaceutique, dans les conditions d'utilisation proposées, sur les espèces non visées (flore et faune). Une exposition unique, prolongée ou répétée peut être à l'origine d'effets réversibles ou irréversibles.
ii) Le choix des organismes non visés appropriés aux fins d'évaluation expérimentale des effets environnementaux doit être fondé sur les informations concernant le micro-organisme comme requis à l'annexe I, partie B, et sur les informations concernant les adjuvants et les autres composants, comme requis aux sections 1 à 9 de la présente annexe. Ces éléments doivent permettre de choisir en vue des tests les organismes appropriés, à savoir par exemple des organismes étroitement apparentés à l'organisme cible.
iii) En particulier, les informations relatives au produit phytopharmaceutique et les autres données pertinentes ainsi que les informations relatives au micro-organisme doivent être suffisantes pour :
- définir les symboles de danger, les mises en garde et les mentions types relatives à la nature des risques et aux conseils de prudence pour la protection de l'environnement à faire figurer sur les emballages (conteneurs) ;
- permettre une évaluation des risques à court terme comme à long terme pour les espèces non visées (populations, communautés et processus, selon le cas) ;
- décider s'il y a lieu de prendre des précautions particulières pour protéger les espèces non visées.
iv) Il y a lieu de mentionner tous les effets potentiellement néfastes constatés au cours des investigations de routine sur les effets environnementaux. Il convient également d'effectuer et de rapporter les études complémentaires qui se révéleraient nécessaires pour identifier les mécanismes en cause et évaluer l'importance des effets constatés.
v) En général, une grande partie des données concernant l'incidence sur les espèces non visées, exigées pour l'agrément des produits phytopharmaceutiques, auront été présentées et évaluées en vue de l'inclusion du/des micro-organisme(s) dans l'annexe I de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques.
vi) S'il est nécessaire de disposer des facteurs de l'exposition pour décider si une étude doit être effectuée, il y a lieu d'utiliser les données obtenues conformément aux dispositions de l'annexe II, partie B, section 9. Toutes les données utiles concernant le produit phytopharmaceutique et le micro-organisme doivent être prises en considération pour l'évaluation de l'exposition. Il conviendra, le cas échéant, d'utiliser les paramètres prévus au présent chapitre. Si les données disponibles indiquent que le produit phytopharmaceutique a un effet plus puissant que le micro-organisme, il convient d'utiliser les données relatives aux effets du produit phytopharmaceutique sur les organismes non visés pour le calcul des rapports effets/exposition importants.
vii) Pour faciliter l'évaluation des résultats obtenus et de leur portée, il y a lieu, dans la mesure du possible, d'utiliser pour les différents tests la même souche de chacune des espèces concernées.
10.1. Effets sur les oiseaux.
Lorsque les informations disponibles sur le micro-organisme ne permettent pas de prédire les effets du produit phytopharmaceutique, il y a lieu de fournir les informations visées à l'annexe I, partie B, section 8, point 8.1, sauf s'il est possible de démontrer que toute exposition des oiseaux est improbable.
10.2. Effets sur les organismes aquatiques.
Lorsque les informations disponibles sur le micro-organisme ne permettent pas de prédire les effets du produit phytopharmaceutique, il y a lieu de fournir les informations visées à l'annexe I, partie B, section 8, point 8.2, sauf s'il est possible de démontrer que toute exposition des organismes aquatiques est improbable.
10.3. Effets sur les abeilles.
Lorsque les informations disponibles sur le micro-organisme ne permettent pas de prédire les effets du produit phytopharmaceutique, il y a lieu de fournir les informations visées à l'annexe I, partie B, section 8, point 8.3, sauf s'il est possible de démontrer que toute exposition des abeilles est improbable.
10.4. Effets sur les arthropodes autres que les abeilles.
Lorsque les informations disponibles sur le micro-organisme ne permettent pas de prédire les effets du produit phytopharmaceutique, il y a lieu de fournir les informations visées à l'annexe I, partie B, section 8, point 8.4, sauf s'il est possible de démontrer que toute exposition des arthropodes autres que les abeilles est improbable.
10.5. Effets sur les vers de terre.
Lorsque les informations disponibles sur le micro-organisme ne permettent pas de prédire les effets du produit phytopharmaceutique, il y a lieu de fournir les informations visées à l'annexe I, partie B, section 8, point 8.5, sauf s'il est possible de démontrer que toute exposition des vers de terre est improbable.
10.6. Effets sur les micro-organismes du sol.
Lorsque les informations disponibles sur le micro-organisme ne permettent pas de prédire les effets du produit phytopharmaceutique, il y a lieu de fournir les informations visées à l'annexe I, partie B, section 8, point 8.6, sauf s'il est possible de démontrer que toute exposition des micro-organismes du sol non visés est improbable.
10.7. Etudes complémentaires.
Un avis d'expert est exigé pour décider s'il convient d'engager des études complémentaires. Cette décision doit prendre en considération les informations disponibles au titre de la présente section mais également d'autres sections, et notamment les données relatives à la spécificité du micro-organisme et aux situations d'exposition prévues. Les observations réalisées lors de tests d'efficacité peuvent également fournir à cet égard des informations utiles. Il y a lieu d'accorder une attention particulière aux organismes utiles dans le cadre de la gestion intégrée des cultures, qu'ils soient présents naturellement ou qu'ils aient été introduits délibérément. Il convient ainsi, notamment, de prendre en considération la compatibilité du produit avec la gestion intégrée des cultures. Les études complémentaires peuvent comprendre des études pointues sur d'autres espèces ou des études à un niveau supérieur consacrées, par exemple, à certains organismes non visés. Avant d'entamer ces études, le demandeur doit obtenir l'accord des autorités compétentes pour le type d'études à effectuer.
11. Synthèse et évaluation des incidences sur l'environnement.
Il convient d'effectuer une synthèse et une évaluation de toutes les données concernant l'incidence sur l'environnement, conformément aux lignes directrices établies par les autorités compétentes des Etats membres au sujet du format de telles synthèses et évaluations. Le document doit comprendre une évaluation critique et détaillée de ces données dans la perspective des lignes directrices et des critères importants pour l'évaluation et la prise de décision, en prêtant une attention particulière aux risques éventuels ou effectifs pour l'environnement et les espèces non visées ainsi qu'à l'importance, à la qualité et à la fiabilité de la base de données. Les thèmes suivants doivent notamment être traités :
- la prédiction de la dissémination et du devenir dans l'environnement, ainsi que les durées correspondantes ;
- l'identification des espèces et des populations non visées susceptibles d'être affectées, et l'ampleur estimée de leur exposition potentielle ;
- l'identification des précautions nécessaires pour éviter ou réduire au minimum la contamination de l'environnement et protéger les espèces non visées.
4. Dans des cas individuels, il peut être nécessaire de demander pour des produits entrant dans la composition de la formulation certaines informations prévues à l'annexe I, partie A.
Préalablement à toute demande d'informations concernant un tel produit, il est procédé à l'examen de toute information mise à la disposition de l'autorité compétente, notamment :
- lorsque l'utilisation du produit est autorisée dans les denrées alimentaires, les matières premières pour aliments du bétail, médicaments ou cosmétiques conformément à la législation communautaire,
ou
- lorsqu'une fiche de données de sécurité a été présentée pour le produit considéré conformément à la directive n° 67/548/C.E.E. du conseil.
Calendrier de mise en œuvre des nouvelles dispositions relatives à l'étiquetage et à l'emballage.
Annexe III
(Arrêté du 28 février 2005, article 1er, annexe II)
Calendrier de mise en oeuvre des nouvelles dispositions relatives à l'étiquetage et à l'emballage
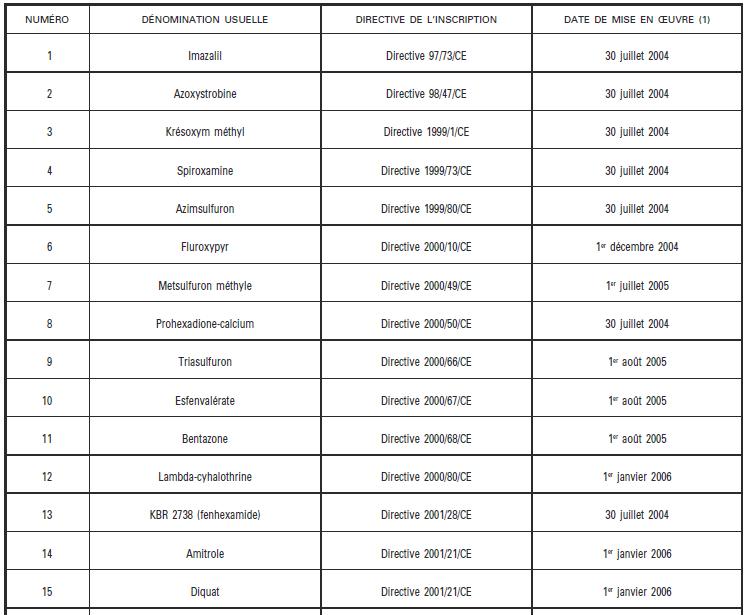
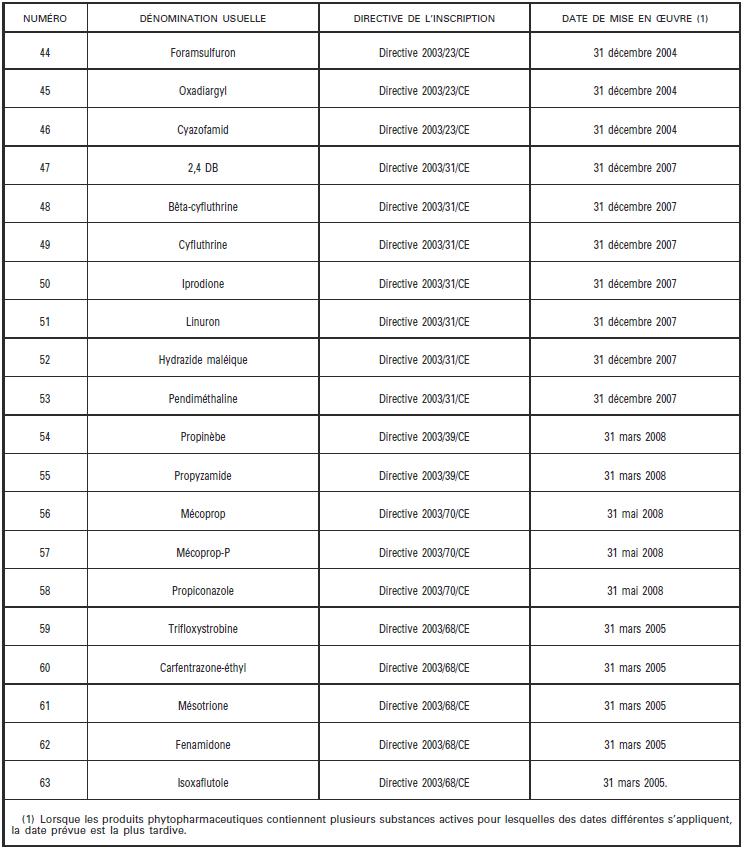
Phrases types indiquant les risques particuliers pour l'homme ou l'environnement visées à l'article 34 (f).
Annexe IV
(Arrêté du 25 février 2005, article 1er annexe I)
Introduction.
Les phrases types supplémentaires ci-après sont définies pour s'ajouter aux phrases prévues par l'arrêté du 9 novembre 2004 définissant les critères de classification et les conditions d'étiquetage et d'emballage des préparations dangereuses qui transpose la directive 1999/45/CE du Parlement européen et du Conseil du 31 mai 1999, et qui s'applique aux produits phytopharmaceutiques contenant des micro-organismes ou des virus en tant que substances actives. L'étiquetage des produits contenant ces substances actives doit également être conforme aux dispositions concernant les essais relatifs à la sensibilisation cutanée et respiratoire figurant aux annexes I-B et II-B de l'arrêté du 6 septembre 1994.
Les phrases harmonisées constituent la base des instructions d'utilisation spécifiques et complémentaires et s'appliquent, sans préjudice, d'autres éléments de l'article 34 (k) à (n) de l'arrêté du 6 septembre 1994 et de l'article 3 du décret du 11 mai 1937 susvisé.
1. Phrases types indiquant les risques particuliers.
1.1. Risques particuliers pour l'homme.
RSh 1 - Toxique par contact oculaire.
RSh 2 - Peut entraîner une photosensibilisation.
RSh 3 - Le contact avec les vapeurs peut provoquer des brûlures de la peau et des yeux ; le contact avec le gaz liquide peut causer des engelures.
1.2. Risques particuliers pour l'environnement, RSe.
Aucun.
2. Critères d'application des phrases types indiquant les risques particuliers.
2.1. Critères d'application des phrases types pour l'homme.
RSh 1 - Toxique par contact oculaire :
La phrase doit être utilisée lorsqu'un essai d'irritation des yeux visé au point 7.1.5 (= sensibilisation de la peau) de l'annexe II A provoque, chez les animaux d'essais, des signes manifestes de toxicité systémique (par exemple liés à l'inhibition de la cholinestérase) ou une mortalité susceptibles d'être attribués à l'absorption de la substance active à travers les muqueuses de l'oeil. La phrase concernant le risque doit également être utilisée lorsque la preuve de la toxicité systémique chez l'homme après contact oculaire est établie.
Il y a lieu de spécifier la protection oculaire dans ces cas, comme indiqué dans les dispositions générales de l'annexe V de l'arrêté du 6 septembre 1994.
RSh 2 - Peut entraîner une photosensibilisation :
La phrase doit être utilisée lorsque des systèmes expérimentaux ou une exposition humaine dûment établie démontrent clairement que les produits exercent un effet photosensibilisant. La phrase doit également être utilisée pour les produits qui contiennent une substance active ou un composant donnés qui exercent un effet photosensibilisant chez l'homme, si cet élément photosensibilisant est présent à une concentration de 1 % ou plus.
Il y a lieu de spécifier les mesures de protection individuelle dans ce cas, comme indiqué dans les dispositions générales de l'annexe V de l'arrêté du 6 septembre 1994.
RSh 3 - Le contact avec les vapeurs peut provoquer des brûlures de la peau et des yeux ; le contact avec le gaz liquide peut causer des engelures :
La phrase doit être utilisée pour les produits phytopharmaceutiques préparés sous la forme de gaz liquéfié, le cas échéant (par exemple les préparations de bromure de méthyle).
Il y a lieu de spécifier les mesures de protection individuelle dans ce cas, comme indiqué dans les dispositions générales de l'annexe V de l'arrêté du 6 septembre 1994.
Dans les cas où R 34 et R 35 s'appliquent conformément à l'arrêté du 9 novembre 2004 transposant la directive 1999/45/CE, la phrase ne doit pas être utilisée.
2.2. Critères d'application des phrases types pour l'environnement.
Aucun.
Phrases types indiquant les précautions à prendre pour la protection de l'homme ou de l'environnement visées à l'article 34 (g).
Annexe V
(Arrêté du 28 février 2005, article 1er, annexe II)
Introduction.
Les phrases types supplémentaires ci-après sont définies pour s'ajouter aux phrases prévues par l'arrêté du 9 novembre 2004 définissant les critères de classification et les conditions d'étiquetage et d'emballage des préparations dangereuses qui transpose la directive 1999/45/CE du Parlement européen et du Conseil du 31 mai 1999, et qui s'applique aux produits phytopharmaceutiques. Les dispositions prévues par cet arrêté sont également utilisées pour les produits phytopharmaceutiques contenant des micro-organismes ou des virus en tant que substances actives. L'étiquetage des produits contenant ces substances actives doit également être conforme aux dispositions concernant les essais relatifs à la sensibilisation cutanée et respiratoire figurant aux annexes I-B et II-B de l'arrêté du 6 septembre 1994.
Les phrases harmonisées constituent la base des instructions d'utilisation spécifiques et complémentaires et s'appliquent sans préjudice d'autres éléments de l'article 34 (k à n) de l'arrêté du 6 septembre 1994 et de l'article 3 du décret du 11 mai 1937 susvisé.
1. Dispositions générales.
Tous les produits phytopharmaceutiques doivent porter un étiquetage mentionnant la phrase ci-après, complétée le cas échéant par le texte entre crochets :
SP 1 - Ne pas polluer l'eau avec le produit ou son emballage. [Ne pas nettoyer le matériel d'application près des eaux de surface. Eviter la contamination via les systèmes d'évacuation des eaux à partir des cours de ferme ou des routes].
2. Précautions spécifiques à prendre.
2.1. Précautions à prendre par les utilisateurs.
2.1.1. Dispositions générales :
2.1.1.1. Un équipement de protection individuelle approprié pour les utilisateurs peut être précisé et des éléments spécifiques dudit équipement peuvent être prescrits (combinaison, tablier, gants, chaussures robustes, bottes en caoutchouc, protection du visage, écran facial, lunettes de protection, chapeau, cagoule ou masque à gaz d'un type spécifié). Ces précautions supplémentaires à prendre s'appliquent sans préjudice des phrases types prévues par l'arrêté du 9 novembre 2004 définissant les critères de classification et les conditions d'étiquetage et d'emballage des préparations dangereuses.
2.1.1.2. Les actions spécifiques qui requièrent un équipement de protection particulier, telles que le mélange, le chargement ou la manipulation du produit non dilué, l'application ou la vaporisation du produit dilué, la manipulation de matières fraîchement traitées, telles que les végétaux ou le sol, ou encore l'accès aux zones fraîchement traitées, peuvent être précisées.
2.1.1.3. Des spécifications relatives aux contrôles techniques peuvent être ajoutées, telles que :
- utiliser un dispositif de versement clos pour transvaser le pesticide de l'emballage du produit dans le réservoir du pulvérisateur ;
- l'utilisateur doit se trouver dans une cabine close [pourvue d'un système de climatisation/de filtration de l'air] lors de la pulvérisation ;
- les contrôles techniques peuvent remplacer l'équipement de protection individuelle s'ils fournissent un degré de protection égal ou supérieur.
2.1.2. Dispositions spécifiques :
SPo 1 - Après contact avec la peau, éliminer d'abord le produit avec un chiffon sec, puis laver la peau abondamment à l'eau.
SPo 2 - Laver tous les équipements de protection après utilisation.
SPo 3 - Après déclenchement de la fumigation, ne pas inhaler la fumée et quitter la zone traitée immédiatement.
SPo 4 - L'emballage doit être ouvert à l'extérieur par temps sec.
SPo 5 - Ventiler [à fond/ou durée à préciser/jusqu'au séchage de la pulvérisation] les zones/serres traitées avant d'y accéder.
2.2. Précautions à prendre pour l'environnement :
SPe 1 - Pourprotéger [les eaux souterraines / les organismes du sol], ne pas appliquer ce produit ou tout autre produit contenant (préciser la substance ou la famille de substances selon le cas) plus de (fréquence à préciser).
SPe 2 - Pour protéger [les eaux souterraines / les organismes aquatiques], ne pas appliquer ce produit sur (type de sol ou situation à préciser).
SPe 3 - Pour protéger [les organismes aquatiques / les plantes non cibles / les arthropodes non cibles / les insectes], respecter une zone non traitée de (distance à préciser) par rapport à [la zone non cultivée adjacente/aux points d'eau].
SPe 4 - Pour protéger [les organismes aquatiques/les plantes non cibles], ne pas appliquer sur des surfaces imperméables telles que le bitume, le béton, les pavés, [les voies ferrées] et dans toute autre situation où le risque de ruissellement est important.
SPe 5 - Pour protéger [les oiseaux/mammifères sauvages], le produit doit être entièrement incorporé dans le sol ; s'assurer que le produit est également incorporé en bout de sillons.
SPe 6 - Pour protéger [les oiseaux/mammifères sauvages], récupérer tout produit accidentellement répandu.
SPe 7 - Ne pas appliquer durant la période de reproduction des oiseaux.
SPe 8 - Dangereux pour les abeilles./ Pour protéger les abeilles et autres insectes pollinisateurs, ne pas appliquer durant la floraison./ Ne pas utiliser en présence d'abeilles./ Retirer ou couvrir les ruches pendant l'application et (indiquer la période) après traitement./ Ne pas appliquer lorsque des adventices en fleur sont présentes./ Enlever les adventices avant leur floraison./ Ne pas appliquer avant (indiquer la date).
2.3. Précautions à prendre dans le cadre des bonnes pratiques agricoles :
SPa 1 - Pour éviter le développement de résistances, ne pas appliquer ce produit ou tout autre contenant (préciser la substance ou la famille de substances selon le cas) plus de (nombre d'applications ou durée à préciser).
2.4. Précautions spécifiques à prendre pour les rodenticides :
SPr 1 - Les appâts doivent être disposés de manière à minimiser le risque d'ingestion par d'autres animaux. Sécuriser les appâts afin qu'ils ne puissent être emmenés par les rongeurs.
SPr 2 - La zone de traitement doit faire l'objet d'un marquage pendant la période de traitement. Le risque d'empoisonnement (primaire ou secondaire) par l'anticoagulant ainsi que son antidote doivent être mentionnés.
SPr 3 - Les rongeurs morts doivent être retirés quotidiennement de la zone de traitement pendant toute la période de traitement. Ne pas les jeter dans les poubelles ni dans les décharges.
3. Critères d'application des phrases types indiquant les précautions spécifiques à prendre.
3.1. Introduction :
Généralement, les produits phytopharmaceutiques ne sont autorisés que pour les usages définis sur la base d'une évaluation conforme aux principes uniformes établis à l'annexe III de l'arrêté du 6 septembre 1994.
Dans la mesure du possible, les précautions spécifiques à prendre doivent tenir compte des résultats desdites évaluations effectuées conformément aux principes uniformes et s'appliquer en particulier dans les cas où des mesures visant à réduire les risques sont nécessaires pour prévenir des effets inacceptables.
3.2. Critères d'application des phrases types indiquant les précautions à prendre par les utilisateurs :
SPo 1 - Après contact avec la peau, éliminer d'abord le produit avec un chiffon sec, puis laver abondamment à l'eau.
La phrase doit être utilisée pour les produits phytopharmaceutiques contenant des composants susceptibles de réagir violemment au contact de l'eau, par exemple les sels de cyanure ou le phosphure d'aluminium.
SPo 2 - Laver tous les équipements de protection après utilisation.
La phrase est recommandée lorsque des vêtements de protection sont nécessaires pour protéger les utilisateurs. Elle est obligatoire pour tous les produits phytopharmaceutiques classés T ou T+.
SPo 3 - Après déclenchement de la fumigation, ne pas inhaler la fumée et quitter la zone traitée immédiatement.
La phrase peut être utilisée pour les produits phytopharmaceutiques utilisés pour la fumigation dans le cas où le port d'un masque n'est pas justifié.
SPo 4 - L'emballage doit être ouvert à l'extérieur par temps sec.
La phrase doit être utilisée pour les produits phytopharmaceutiques contenant des substances actives susceptibles de réagir violemment au contact de l'eau ou de l'air humide, par exemple le phosphure d'aluminium, ou sujettes à l'inflammation spontanée, telles que les dithiocarbamates (alkylène-bis). La phrase peut également être utilisée pour les produits volatils classés R 20, R 23 ou R 26. L'avis d'experts doit être pris en considération dans les cas individuels afin d'évaluer si les propriétés de la préparation et l'emballage sont de nature à présenter des risques pour l'utilisateur.
SPo 5 - Ventiler [à fond/ou durée à préciser/jusqu'au séchage de la pulvérisation] les zones/serres traitées avant d'y accéder.
La phrase peut être utilisée pour les produits phytopharmaceutiques employés dans les serres ou dans les autres espaces confinés tels que les entrepôts.
3.3. Critères d'application des phrases types indiquant les précautions à prendre pour l'environnement :
SPe 1 - Pour protéger [les eaux souterraines/les organismes du sol], ne pas appliquer ce produit ou tout autre produit contenant [préciser la substance ou la famille de substances selon le cas] plus de [fréquence à préciser].
La phrase doit être utilisée pour les produits phytopharmaceutiques pour lesquels une évaluation conforme aux principes uniformes démontre, pour l'un ou plusieurs des usages désignés, que des mesures visant à réduire les risques sont nécessaires pour éviter l'accumulation dans le sol, les effets sur les vers de terre ou autres organismes vivant dans le sol, la microflore du sol et/ou la contamination des eaux souterraines.
SPe 2 - Pour protéger [les eaux souterraines/les organismes aquatiques], ne pas appliquer ce produit sur [type de sol ou situation à préciser].
La phrase doit être utilisée en tant que mesure visant à réduire les risques pour prévenir la contamination des eaux souterraines ou des eaux de surface exposées (par exemple en association avec le type de sol, la topographie ou pour les sols drainés), si une évaluation conforme aux principes uniformes démontre, pour l'un ou plusieurs des usages désignés, que des mesures visant à réduire les risques sont nécessaires pour éviter des effets inacceptables.
SPe 3 - Pour protéger [les organismes aquatiques/les plantes non cibles/les arthropodes non cibles/les insectes], respecter une zone non traitée de [distance à préciser] par rapport à la [zone non cultivée adjacente/aux points d'eau].
La phrase doit être utilisée pour protéger les plantes non cibles, les arthropodes non cibles et/ou les organismes aquatiques si une évaluation conforme aux principes uniformes démontre, pour l'un ou plusieurs des usages désignés, que des mesures visant à réduire les risques sont nécessaires pour éviter des effets inacceptables.
SPe 4 - Pour protéger [les organismes aquatiques/les plantes non cibles], ne pas appliquer sur des surfaces imperméables telles que le bitume, le béton, les pavés, [les voies ferrées] et dans toute autre situation où le risque de ruissellement est important.
En fonction du type d'utilisation du produit phytopharmaceutique, la phrase peut être utilisée pour prévenir le risque de ruissellement et protéger les organismes aquatiques ou les plantes non cibles.
SPe 5 - Pour protéger [les oiseaux/les mammifères sauvages], le produit doit être incorporé dans le sol ; s'assurer que le produit est également incorporé en bout de sillons.
La phrase peut être utilisée pour les produits phytopharmaceutiques tels que les granules ou les pastilles qui doivent être incorporés dans le sol pour protéger les oiseaux ou les mammifères sauvages.
SPe 6 - Pour protéger [les oiseaux/les mammifères sauvages], récupérer tout produit accidentellement répandu.
La phrase doit être utilisée pour les produits phytopharmaceutiques tels que les granules ou les pastilles, pour éviter leur absorption par les oiseaux ou les mammifères sauvages. Elle est recommandée pour toutes les préparations solides utilisées non diluées.
SPe 7 - Ne pas appliquer durant la période de reproduction des oiseaux.
La phrase doit être utilisée lorsqu'une évaluation conforme aux principes uniformes démontre, pour l'un ou plusieurs des usages désignés, qu'une telle mesure visant à réduire les risques est nécessaire.
SPe 8 - Dangereux pour les abeilles./Protéger les abeilles et autres insectes pollinisateurs, ne pas appliquer durant la floraison./Ne pas utiliser en présence d'abeilles./Retirer ou couvrir les ruches pendant l'application et [indiquer la période] après traitement./ Ne pas appliquer lorsque de adventices en fleur sont présentes./ Enlever les adventices avant leur floraison./Ne pas appliquer avant [indiquer la date].
La phrase doit être utilisée dans le cas des produits phytopharmaceutiques pour lesquels une évaluation conforme aux principes uniformes démontre, pour l'un ou plusieurs des usages désignés, que des mesures visant à réduire les risques sont nécessaires pour protéger les abeilles ou d'autres insectes pollinisateurs. En fonction du type d'utilisation du produit phytopharmaceutique et d'autres dispositions règlementaires nationales pertinentes, la formulation appropriée sera choisie pour réduire le risque pour les abeilles et autres insectes pollinisateurs ainsi que pour leur couvain.
3.4. Critères d'application des phrases types indiquant les précautions à prendre dans le cadre des bonnes pratiques agricoles :
SPa 1 - Pour éviter le développement de résistances, ne pas appliquer ce produit ou tout autre contenant [préciser la substance ou la famille de substances selon le cas] plus de [nombre d'applications ou durée à préciser].
La phrase doit être utilisée lorsqu'une telle restriction apparaît nécessaire pour limiter le risque de développement de résistances.
3.5. Critères d'application des phrases types indiquant les précautions à prendre pour les rodenticides :
SPr 1 - Les appâts doivent être disposés de manière à minimiser le risque d'ingestion par d'autres animaux. Sécuriser les appâts afin qu'ils ne puissent être emmenés par les rongeurs.
Pour assurer son respect par les utilisateurs, la phrase doit figurer d'une manière visible sur l'étiquette de façon à prévenir les utilisations erronées dans toute la mesure du possible.
SPr 2 - La zone de traitement doit faire l'objet d'un marquage pendant la période de traitement. Le risque d'empoisonnement (primaire ou secondaire) par l'anticoagulant ainsi que son antidote doivent être mentionnés.
La phrase doit figurer d'une manière visible sur l'étiquette de façon à prévenir les empoisonnements accidentels dans toute la mesure du possible.
SPr 3 - Les rongeurs morts doivent être retirés quotidiennement de la zone de traitement pendant toute la période de traitement. Ne pas les jeter dans les poubelles ni les décharges.
Afin de prévenir l'empoisonnement secondaire des animaux, la phrase doit être utilisée pour tous les rodenticides contenant des anticoagulants comme substances actives.