(publiée sur le site circulaires.legifrance.gouv.fr)
| Destinataires |
Mmes et MM. les Directeurs des DIRECCTE et des DIECCTE,
Mmes et MM. les Directeurs départementaux chargés de la Protection des Populations,
Mmes et MM. les responsables des services centraux et des services à compétence nationale de la DGCCRF. |
Objet : Réglementation applicable à la mise sur le marché et l’utilisation des produits phytopharmaceutiques.
| Résumé : Les conditions de mise sur le marché et d’utilisation des produits phytopharmaceutiques sont encadrées depuis le 14 juin 2011 par le règlement (CE) n°1107/2009 concernant la mise sur le marché des produits phytopharmaceutiques et les textes de transposition de la directive 2009/128/CE instaurant un cadre d’action communautaire pour parvenir à une utilisation des pesticides compatible avec le développement durable. La présente note a pour objectif de présenter le nouveau dispositif réglementaire et les principales obligations, en termes d’autorisation, d’étiquetage, d’emballage, de conditions de vente et d’utilisation des produits. |
L’utilisation de produits phytopharmaceutiques constitue l’un des moyens les plus importants pour protéger les végétaux et produits végétaux contre les organismes nuisibles, y compris les mauvaises herbes, et pour améliorer la production agricole.
Les produits phytopharmaceutiques peuvent cependant également avoir des effets non bénéfiques sur la production végétale. L’utilisation de ces produits peut présenter des risques pour l’homme, les animaux et l’environnement, notamment s’ils sont mis sur le marché sans avoir été officiellement testés et autorisés et s’ils sont utilisés d’une manière incorrecte.
La révision de la directive 91/414/CEE (1), a abouti en 2009 à la publication du règlement (CE) n°1107/2009 (2) et de la directive 2009/128/CE (3), constituant deux des quatre textes du « paquet pesticide (4) ». Ces textes modernisent les conditions de mise sur le marché et d’utilisation des produits phytopharmaceutiques. Les adaptations du droit national en relation avec ces deux textes ont abouti à la refonte des parties législative et réglementaire du code rural et de la pêche maritime relatives aux produits phytopharmaceutiques. L’ordonnance n°2011-840 du 15 juillet 2011 a modifié les chapitres III, IV et VI du titre V du livre II (partie législative). Les décrets n° 2011-1325 du 18 octobre 2011, n° 2011-1650 du 27 novembre 2011 et n° 2012-755 du 9 mai 2012 ont réécrit les chapitres III et IV du titre V du livre II (partie réglementaire).
Cette note a pour objectif de présenter les dispositions essentielles européennes et françaises qui encadrent la mise sur le marché et l’utilisation des produits phytopharmaceutiques.
Le Sous-Directeur
Jean-Louis Gérard
(1) Directive 91/414/CEE du Conseil du 15 juillet 1991 concernant la mise sur le marché des produits phytopharmaceutiques.
(2) Règlement (CE) n°1107/2009 du Parlement européen et du Conseil du 21 octobre 2009 concernant la mise sur le marché des produits phytopharmaceutiques et abrogeant les directives 79/117/CEE et 91/414/CEE du Conseil
(3) Directive 2009/128/CE du Parlement européen et du Conseil du 21 octobre 2009 instaurant un cadre d’action communautaire pour parvenir à une utilisation des pesticides compatible avec le développement durable
(4) Le « paquet pesticide » comporte également le règlement (CE) n° 1185/2009 du Parlement européen et du Conseil du 25 novembre 2009 relatif aux statistiques sur les pesticides ainsi que le règlement (CE) n° 396/2005 du Parlement européen et du Conseil du 23 février 2005 concernant les limites maximales applicables aux résidus de pesticides présents dans ou sur les denrées alimentaires et les aliments pour animaux d'origine végétale et animale et modifiant la directive 91/414/CEE du Conseil.
I. Présentation des principaux textes encadrant la mise sur le marché et l’utilisation des produits phytopharmaceutiques
I.1 Mise sur le marché des produits phytopharmaceutiques :
- Le règlement (CE) n° 1107/2009 du Parlement européen et du Conseil du 21 octobre 2009 concernant la mise sur le marché des produits phytopharmaceutiques et abrogeant les directives 79/117/CEE et 91/414/CEE du Conseil est entré en application au 14 juin 2011. Les dispositions sont organisées en onze chapitres.
Chapitre I : dispositions générales (définitions, champ d’application),
Chapitre II : substances actives, phytoprotecteurs, synergistes et coformulants,
Chapitre III : produits phytopharmaceutiques
Chapitre IV : adjuvants
Chapitre V : protection et partage des données
Chapitre VI : accès du public à l’information
Chapitre VII : emballage, étiquetage et publicité
Chapitre VIII : contrôles
Chapitre IX : situation d’urgence
Chapitre X : dispositions administratives et financières
Chapitre XI : dispositions transitoires et finales
Ce règlement est accompagné de 5 règlements d’exécution :
- Le règlement d’exécution (UE) n°540/2011 modifié (5) de la Commission du 25 mai 2011 portant application du règlement (CE) n°1107/2009 du Parlement européen et du Conseil en ce qui concerne la liste des substances actives approuvées.
- Le règlement (UE) n° 544/2011 de la Commission du 10 juin 2011 portant application du règlement (CE) n°1107/2009 du Parlement européen et du Conseil en ce qui concerne les exigences en matière de données applicables aux substances actives.
- Le règlement (UE) n° 545/2011 de la Commission du 10 juin 2011 portant application du règlement (CE) n°1107/2009 du Parlement européen et du Conseil en ce qui concerne les exigences en matière de données applicables aux produits phytopharmaceutiques.
- Le règlement (UE) n° 546/2011 de la Commission du 10 juin 2011 portant application du règlement (CE) n°1107/2009 du Parlement européen et du Conseil en ce qui concerne les principes uniformes d’évaluation et d’autorisation des produits phytopharmaceutiques.
- Le règlement (UE) n° 547/2011 de la Commission du 8 juin 2011 portant application du règlement (CE) n°1107/2009 du Parlement européen et du Conseil concernant les exigences en matière d’étiquetage de produits phytopharmaceutiques.
(5) Modifications : règlements d’exécution (UE) n°541/2011 et n°542/2011 de la Commission du 1er juin 2011
I.2 Utilisation des produits phytopharmaceutiques :
- La directive 2009/128/CE du Parlement européen et du Conseil du 21 octobre 2009 instaurant un cadre d’action communautaire pour parvenir à une utilisation des pesticides compatible avec le développement durable. Les dispositions sont organisées en six chapitres.
Chapitre I : dispositions générales (définitions, champ d’application, plans d’actions nationaux),
Chapitre II : formation, vente de pesticides, information et sensibilisation,
Chapitre III : matériel d’application des pesticides,
Chapitre IV : pratiques et utilisations spécifiques (pulvérisation aérienne, mesures spécifiques de protection du milieu aquatique et de l’eau potable, réduction de l’utilisation des pesticides ou des risques dans des zones spécifiques, manipulation et stockage des pesticides et traitement de leurs emballages et des restes de produits, lutte intégrée contre les ennemis des cultures),
Chapitre V : indicateurs, rapports et échanges d’informations,
Chapitre VI : dispositions finales
Les dispositions nationales ont été mises en conformité avec les règlements et la directive précités au moyen de :
- L’ordonnance n° 2011-840 du 15 juillet 2011 relative à la mise en conformité des dispositions nationales avec le droit de l’Union européenne sur la mise sur le marché et l’utilisation des produits phytopharmaceutiques qui a notamment réécrit le chapitre III (partie législative) et modifié les chapitres IV et VI (partie législative) du titre V du livre II du code rural et de la pêche maritime.
- Le décret n° 2011-1325 du 18 octobre 2011 fixant les conditions de délivrance, de renouvellement, de suspension et de retrait des agréments des entreprises et des certificats individuels pour la mise en vente, la distribution à titre gratuit, l’application et le conseil à l’utilisation des produits phytopharmaceutiques, qui a réécrit le chapitre IV (partie réglementaire) du titre V du livre II du code rural et de la pêche maritime.
- Le décret n° 2011-1650 du 25 novembre 2011 relatif aux modalités de déclaration et de reversement de la redevance pour pollutions diffuses et aux modalités de tenue des registres mentionnés aux articles L.254-3-1 et L.254-6 du code rural et de la pêche maritime, qui a modifié le chapitre IV (partie réglementaire) du titre V du livre II du code rural et de la pêche maritime.
- Le décret n° 2012-755 du 9 mai 2012 relatif à la mise en conformité des dispositions nationales avec le droit de l’Union européenne en ce qui concerne la mise sur le marché et l’utilisation des produits phytopharmaceutiques, qui a réécrit le chapitre III (partie réglementaire) du titre V du livre II du code rural et de la pêche maritime [entrée en vigueur le 1er juillet 2012].
A cela s’ajoutent de nombreux arrêtés d’application permettant d’encadrer la mise sur le marché des produits phytopharmaceutiques destinés au grand public, le traitement aérien, l’utilisation (principes généraux et mélanges extemporanés), les conditions d’agrément et de certification des distributeurs, conseillers et applicateurs de produits phytopharmaceutiques. Ces arrêtés sont cités en tant que de besoin dans les paragraphes suivants.
II. Définition d’un produit phytopharmaceutique
Les produits phytopharmaceutiques sont définis, à l’article 2 du RCE n° 1107/2009 comme étant « les produits, sous la forme dans laquelle ils sont livrés à l’utilisateur, composés de substances actives, phytoprotecteurs ou synergistes, ou en contenant, et destinés à l’un des usages suivants :
a) protéger les végétaux ou les produits végétaux contre tous les organismes nuisibles ou prévenir l’action de ceux-ci, sauf si ces produits sont censés être utilisés principalement pour des raisons d’hygiène plutôt que pour la protection des végétaux ou des produits végétaux ;
b) exercer une action sur les processus vitaux des végétaux, telles les substances, autres que les substances nutritives, exerçant une action sur leur croissance ;
c) assurer la conservation des produits végétaux, pour autant que ces substances ou produits ne fassent pas l’objet de dispositions communautaires particulières concernant les agents conservateurs ;
d) détruire les végétaux ou les parties de végétaux indésirables, à l’exception des algues à moins que les produits ne soient appliqués sur le sol ou l’eau pour protéger les végétaux ;
e) freiner ou prévenir une croissance indésirable des végétaux, à l’exception des algues à moins que les produits ne soient appliqués sur le sol ou l’eau pour protéger les végétaux. »
Ainsi, ces différents usages correspondent par exemple aux insecticides (a), fongicides (a), acaricides (a), nématicides (a), répulsifs (a) ; à certains stimulateurs de croissance (b) ou régulateurs de croissance (e) ; aux herbicides (d) et aux produits de traitement après récolte des grains, des agrumes, des ensilages (c).
Il est important de noter que le point commun des produits phytopharmaceutiques est leur action de protection des végétaux ou des produits végétaux contre des organismes qui leur sont nuisibles.
Ce point commun les distingue notamment des produits biocides (6) dont l’objectif est la lutte contre des organismes nuisibles à l’homme, ses activités, ses produits, les animaux ou l’environnement de manière générale.
Il est nécessaire de préciser que l’utilisateur, s’entend comme l’utilisateur « final » du produit, c’est-à-dire la personne qui effectue le traitement à l’aide du produit phytopharmaceutique, qu’il s’agisse d’un professionnel des secteurs agricoles ou non-agricoles (par exemple : espaces verts, golfs) ou d’un amateur souhaitant traiter son potager, son jardin ou encore ses plantes d’intérieur ou de balcon.
Les articles 2 et 3 du RCE n° 1107/2009 explicitent plusieurs termes utilisés dans la définition d’un produit phytopharmaceutique :
- Les substances sont des « éléments chimiques et leurs composés tels qu’ils se présentent à l’état naturel ou tels qu’ils sont produits par l’industrie, y compris toute impureté résultant inévitablement du procédé de fabrication ».
La notion de substance « active » n’est pas définie dans ces textes. Néanmoins, « l’action phytopharmaceutique » d’une substance s’interprète comme tout moyen autre qu’une simple action physique ou mécanique (piège, barrière). Le mode d’action est plus généralement chimique ou biologique.
- Les phytoprotecteurs sont des « substances ou préparations, ajoutées à un produit phytopharmaceutique pour annihiler ou réduire les effets phytotoxiques du produit phytopharmaceutique sur certaines plantes ».
- Les synergistes sont des « substances ou préparations qui, bien que n’ayant pas ou guère d’activité phytopharmaceutique, peuvent renforcer l’activité de la ou des substances actives présentes dans un produit phytopharmaceutique ». A titre d’exemple, le pipéronyl butoxyde est un synergiste.
- Les coformulants sont des « substances ou préparations utilisées ou destinées à être utilisées dans un produit phytopharmaceutique ou un adjuvant, mais qui ne sont ni des substances actives ni des phytoprotecteurs ou synergistes ». Les coformulants assurent la stabilité du produit ; il peut s’agir d’émulsifiant, d’agent de mise en suspension, d’antimottant.
- Les végétaux sont « les plantes vivantes et les parties vivantes de plantes, y compris les fruits et légumes frais et les semences ».
- Les produits végétaux sont « les produits d’origine végétale non transformés ou ayant subi une préparation simple telle que mouture, séchage ou pression, pour autant qu’il ne s’agisse pas de végétaux ». La farine, les fleurs coupées sont des produits végétaux.
- Les organismes nuisibles sont des « espèce, souche ou biotype appartenant au règne animal ou au règne végétal ou agent pathogène nuisible aux végétaux ou aux produits végétaux ».
Enfin, il est nécessaire d’ajouter qu’au sens du RCE n° 1107/2009, la mise sur le marché s’entend comme la détention en vue de la vente à l’intérieur de l’Union européenne, y compris l’offre en vue de la vente ou toute autre forme de cession, à titre gratuit ou onéreux, ainsi que la vente, la distribution et les autres formes de cession proprement dites, sauf la restitution au vendeur précédent.
La mise en libre pratique sur le territoire de l’Union constitue une mise sur le marché.
(6) Les produits biocides sont réglementés aux articles L.522-1 et suivants du code de l’environnement
L’objectif de la réglementation européenne étant de garantir un niveau élevé de protection de la santé humaine et animale et de l’environnement, des exigences sont fixées pour les substances entrant dans la composition des produits phytopharmaceutiques.
III.1 les substances actives doivent avoir été approuvées au niveau européen
Les substances actives autorisées dans les produits phytopharmaceutiques sont approuvées au niveau européen, sous réserve qu’elles présentent un intérêt manifeste pour la production végétale et qu’elles n’aient pas d’effets nocifs sur la santé humaine ou animale ou d’effet inacceptable sur l’environnement.
L’article 4 du RCE n° 1107/2009 détermine précisément les critères d’approbation des substances
actives, qui portent notamment sur les effets de ces substances dans les produits phytopharmaceutiques ainsi que sur les effets des résidus de ces substances.
Les exigences en matière de données sont fixées par le règlement (CE) n° 544/2011. Certaines conditions ou restrictions, telles que le degré de pureté minimal de la substance, la nature des impuretés, peuvent être fixées lors de l’approbation (article 6 du RCE n° 1107/2009).
La demande d’approbation ou de modification des conditions d’approbation est introduite par le producteur ou une association de producteurs de la substance active auprès d’un Etat membre, dénommé « Etat membre rapporteur », qui est chargé de l’examiner.
Le dossier de demande d’approbation comprend notamment des informations relatives à une ou plusieurs utilisations représentatives sur une culture très répandue d’au moins un produit phytopharmaceutique contenant la substance active.
A compter de la recevabilité du dossier, l’Etat membre rapporteur dispose de douze mois pour établir et transmettre à la Commission européenne (DGSANCO) et à l’Autorité européenne de sécurité des aliments (dénommé ci-après Autorité européenne, www.efsa.europa.eu), son projet de rapport d’évaluation, évaluant si la substance active est susceptible de satisfaire aux critères d’approbation. Ce projet de rapport d’évaluation comprend notamment des mesures d’atténuation des risques.
L’Autorité européenne communique ce projet de rapport d’évaluation au demandeur et aux autres Etats membres. Elle le met à disposition du public, autorise la présentation d’observations écrites et organise, s’il y a lieu, une consultation d’experts. A l’issue de cette période, l’Autorité européenne adopte des conclusions dans lesquelles elle précise si la substance active est susceptible de satisfaire aux conditions d’approbation.
Dans les six mois à compter de la réception des conclusions de l’Autorité européenne, la Commissioneuropéenne présente un rapport d’examen et un projet de règlement tenant compte du projet de rapport d’évaluation de l’Etat membre rapporteur et des conclusions de l’Autorité européenne.
Le règlement prévoit selon le cas que :
- la substance active est approuvée, sous réserve s’il y a lieu de conditions ou de restrictions ;
- la substance active n’est pas approuvée ;
- les conditions de l’approbation sont modifiées ;
Les substances actives approuvées sont inscrites à l’annexe du règlement (CE) n° 540/2011 modifié. Le site internet de la Commission européenne « EU PESTICIDE DATABASE (7) » permet de connaître le statut effectif de chaque substance active (approuvée, non approuvée) et renvoie sur les documents législatifs correspondants.
Les substances actives sont approuvées pour une durée n’excédant pas dix ans. Sur demande, l’approbation d’une substance active peut être renouvelée pour une période n’excédant pas quinze ans.
Les substances actives approuvées peuvent être qualifiées, le cas échéant, de « substances actives à faible risque », « substances actives de base » ou « substances actives dont on envisage la substitution ».
- Les substances actives à faible risque :
L’annexe II du règlement (RCE) 1107/2009 prévoit qu’une substance active n’est pas considérée comme « à faible risque » si :
→ elle est classée dans au moins une des catégories suivantes : cancérogène, mutagène, toxique pour la reproduction, sensibilisant, très toxique ou toxique, explosive, corrosive.
Ou encore si :
→ elle est persistante (durée de demi-vie dans le sol supérieure à 60 jours), ou
→ le facteur de bioconcentration est supérieur à 100, ou
→ elle est réputée être un perturbateur endocrinien, ou
→ elle a des effets neurotoxiques ou immunotoxiques.
Les substances actives à faible risque sont approuvées pour une durée n’excédant pas quinze ans s’il est prévisible que les produits phytopharmaceutiques contenant cette substance ne présenteront qu’un faible risque pour la santé humaine, la santé animale et l’environnement.
- Les substances actives de base
L’article 23 du RCE n°1107/2009 prévoit qu’une substance active de base :
→ n’est pas une substance préoccupante (8) ;
→ et n’est pas intrinsèquement capable de provoquer des effets perturbateurs sur le système endocrinien, des effets neurotoxiques ou des effets immunotoxiques ;
→ et dont la destination principale n’est pas d’être utilisée à des fins phytosanitaires, mais est néanmoins utile dans la protection phytosanitaire, soit directement, soit dans un produit constitué par la substance et un simple diluant ;
→ et n’est pas mise sur le marché en tant que produit phytopharmaceutique.
Une denrée alimentaire, au sens de l’article 2 du règlement (CE) n°178/2002 est considérée comme une substance de base.
Les substances de base sont approuvées pour une période illimitée.
Les demandes d’approbation sont introduites auprès de la Commission européenne par un Etat membre rapporteur ou toute partie intéressée.
- Les substances actives dont on envisage la substitution
L’annexe II du règlement (RCE) 1107/2009 prévoit qu’une substance active est une substance dont on envisage la substitution si l’une des conditions suivantes est remplie :
→ la dose journalière admissible (DJA), le niveau acceptable d’exposition de l’opérateur ou la dose aiguë de référence (DARf) qui s’y rapportent sont sensiblement inférieurs à ceux de la majorité des substances actives approuvées dans les groupes de substances/catégories d’utilisation ;
→ elle satisfait à deux des critères prévus pour être considérée comme une substance PBT (persistante, bioaccumulable et toxique) ;
→ elle suscite des préoccupations liées à la nature des effets critiques (tels que des effets neurotoxiques ou immunotoxiques pour le développement) qui, combinés aux modes d’utilisation et d’exposition concernés, créent des situations d’utilisation qui restent inquiétantes, par exemple un potentiel élevé de risque pour les eaux souterraines ; même lorsqu’elles s’accompagnent de mesures de gestion des risques très restrictives (équipements de protection individuelle, zones tampons très étendues, etc.) ;
→ elle contient un pourcentage important d’isomères non actifs ;
→ elle est ou doit être classée carcinogène de catégorie 1A ou 1B conformément aux dispositions du règlement (CE) n° 1272/2008 ;
→ elle est ou doit être classée toxique pour la reproduction de catégorie 1A ou 1B conformément aux dispositions du règlement (CE) n° 1272/2008 ;
→ si, sur la base de l’évaluation d’essais fondés sur des lignes directrices adoptées au niveau communautaire ou international ou d’autres données et informations disponibles, examinées par l’Autorité européenne, elle n’est pas considérée comme ayant des effets perturbateurs endocriniens pouvant être néfastes pour l’homme.
Les substances actives dont on envisage la substitution sont approuvées pour une période n’excédant pas sept ans.
(7) http://ec.europa.eu/sanco_pesticides/public/index.cfm
(8) Selon l’article 3 du RCE n°1107/2009 : une substance préoccupante est une substance intrinsèquement capable de provoquer un effet néfaste pour l’homme, les animaux ou l’environnement et contenue ou produite dans un produit phytopharmaceutique à une concentration suffisante pour risquer de provoquer un tel effet. Les substances préoccupantes comprennent, sans se limiter à celles-ci, les substances satisfaisant aux critères fixés pour être classées dangereuses conformément au règlement (CE) n° 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l’étiquetage et à l’emballage des substances et des mélanges et contenues dans le produit phytopharmaceutique à une concentration justifiant que le produit soit considéré comme dangereux au sens de l’article 3 de la directive 1999/45/CE.
Les substances synergistes ou phytoprotecteurs sont soumises aux mêmes conditions d’approbation que celles prévues pour les substances actives (articles 4 à 21 du RCE n° 1107/2009).
Au plus tard au 14 décembre 2014, un règlement d’application établira un programme de travail pour le réexamen progressif des synergistes et phytoprotecteurs se trouvant déjà sur le marché à cette date.
Les coformulants inacceptables sont définis par l’article 27 du RCE n° 1107/2009. Un coformulant inacceptable est interdit dans un produit phytopharmaceutique s’il a été établi :
- que ses résidus résultant d’une application conforme aux bonnes pratiques phytosanitaires dans des conditions réalistes d’utilisation ont un effet nocif sur la santé humaine ou animale ou les eaux souterraines, ou un effet inacceptable sur l’environnement ;
- ou que son utilisation, dans des conditions d’application conformes aux bonnes pratiques phytosanitaires et dans des conditions réalistes d’utilisation, a un effet nocif sur la santé humaine ou animale ou un effet inacceptable sur les végétaux, les produits végétaux ou l’environnement.
Les coformulants inacceptables seront inscrits à l’annexe III du RCE n° 1107/2009.
Au niveau français, lorsque la France est Etat membre rapporteur, l’évaluation est réalisée par l’Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail (Anses).
Les dossiers d’évaluation comprenant notamment les spécifications, l’origine industrielle ainsi que les méthodes d’analyse des substances sont conservés par l’Anses.
IV. Exigences relatives à l’autorisation de mise sur le marché des produits phytopharmaceutiques
Selon l’article 28 du RCE n° 1107/2009, un produit phytopharmaceutique ne peut être mis sur le marché ou utilisé que s’il a été autorisé dans l’Etat membre concerné.
IV.1 Conditions d’octroi d’une autorisation de mise sur le marché
Un produit phytopharmaceutique ne peut être autorisé que si, selon les principes uniformes, il satisfait aux exigences suivantes :
- ses substances actives, phytoprotecteurs et synergistes ont été approuvées ;
Sous certaines conditions prévues à l’article 30 du RCE n° 1107/2009 et jusqu’au 14 juin 2016, les Etats membres peuvent autoriser, pour une période transitoire ne dépassant pas trois ans, la mise sur le marché de produits contenant une substance active qui n’a pas encore été approuvée.
- ses coformulants ne figurent pas dans l’annexe III (coformulants inacceptables) ;
- sa formulation technique est telle que l’exposition de l’utilisateur ou d’autres risques sont limités dans la mesure du possible sans compromettre le fonctionnement du produit ;
- dans l’état actuel des connaissances scientifiques et techniques : il est suffisamment efficace, il n’a pas d’effet nocif sur la santé humaine, sur la santé animale ou d’effets sur le lieu de travail ou sur les eaux souterraines, il n’a aucun effet inacceptable sur les végétaux ou les produits végétaux, il ne provoque ni souffrance, ni douleur inutile chez les animaux vertébrés à combattre et il n’a pas d’effet inacceptable sur l’environnement ;
- la nature et la quantité de ses substances actives, phytoprotecteurs et synergistes et, le cas échéant, les impuretés et coformulants importants sur le plan toxicologique, écotoxicologique ou environnemental peuvent être déterminés à l’aide de méthodes appropriées ;
- les résidus résultant des utilisations autorisées peuvent être déterminés à l’aide de méthodes appropriées d’usage courant dans tous les Etats membres ;
- ses propriétés physico-chimiques ont été déterminées et jugées acceptables pour assurer une utilisation et un stockage adéquats du produit ;
- pour les végétaux ou produits végétaux devant, le cas échéant, être utilisés comme cultures fourragères ou vivrières, les limites maximales de résidus applicables aux produits agricoles concernés par l’utilisation visée dans l’autorisation ont été établies ou modifiées conformément au règlement (CE) n° 396/2005.
L’article 38 du règlement (CE) n° 1107/2009 prévoit la possibilité pour le titulaire de l’AMM d’un produit phytopharmaceutique, de changer l’origine, le procédé de fabrication et / ou le lieu de fabrication de la substance active, du phytoprotecteur ou du synergiste.
Dans ce cas, l’Anses procède à l’évaluation de l’équivalence entre les deux origines, procédés ou lieux de fabrication. Il est alors vérifié que :
- la nouvelle spécification de la substance ne s’écarte pas sensiblement de la spécification figurant dans le règlement approuvant ladite substance ;
- et la substance n’a pas davantage d’effets nocifs dus à ses impuretés que si elle avait été produite selon le procédé de fabrication initialement indiqué dans le dossier étayant l’approbation.
IV.2 Contenu de l’AMM
Selon l’article 31 du RCE n° 1107/2009, l’autorisation de mise sur le marché définit les végétaux ou produits végétaux ainsi que les zones non agricoles (voies ferrées, zones publiques, lieux de stockage) sur lesquelles le produit peut être utilisé et les fins d’une telle utilisation.
Elle énonce la durée de l’autorisation, la classification de danger du produit en vigueur à la date d’édition de l’autorisation, en application de la réglementation (9) sur les substances et mélanges dangereux, ainsi que les exigences relatives à la mise sur le marché et l’utilisation, parmi lesquelles les conditions d’emploi et, le cas échéant :
- la dose maximale par hectare pour chaque utilisation ;
- le nombre maximal d’utilisations par an ;
- le délai entre les utilisations ;
- le délai de rentrée du travailleur après traitement ;
- le délai à respecter entre la dernière utilisation et la récolte ;
- le délai entre la dernière utilisation et la consommation du végétal ou du produit végétal ;
- les restrictions éventuelles à la distribution et à l’emploi du produit ; ces restrictions sont alors indiquées sur l’étiquette ;
- l’obligation d’aviser, avant l’utilisation du produit, tout voisin qui est susceptible d’être exposé à la dérive de pulvérisation et a demandé à être informé ;
- des indications relatives à l’utilisation appropriée conformément aux principes de lutte intégrée contre les ennemis des cultures ;
- la désignation de catégories d’utilisateurs, tels les professionnels et les non-professionnels ;
- l’étiquetage approuvé ;
- la taille de l’emballage et les matériaux qui le composent.
IV.3 Procédure d’obtention, de renouvellement, de modification et de retrait de l’autorisation
Les demandes d’obtention, de renouvellement ou de modification de l’autorisation de mise sur le marché sont déposées auprès de chaque Etat membre dans lequel le produit est destiné à être mis sur le marché (article 33 du RCE n° 1107/2009).
→ La demande d’autorisation de mise sur le marché précise les utilisations envisagées dans chacune des zones (voir IV.9) des Etats membres dans lesquels un dossier a été déposé.
Le dossier est examiné généralement par l’Etat membre proposé par la société. Les Etats membres de la zone dans laquelle la demande a été introduite coopèrent afin d’assurer une répartition équitable de la charge de travail. Ils s’abstiennent de donner suite au dossier tant que l’Etat membre examinant la demande n’a pas réalisé l’évaluation.
L’Etat membre examinant la demande procède à une évaluation indépendante, objective et transparente. Il applique les principes uniformes d’évaluation. Il donne à tous les Etats membres de la même zone, la possibilité de faire part de leurs observations. Il établit un rapport d’évaluation qu’il met à disposition des autres Etats membres de la même zone.
Chaque Etat membre de la zone accorde ou refuse l’autorisation de mise sur le marché sur son territoire, sur la base de ce rapport d’évaluation. L’Etat membre peut mettre en place des mesures nationales d’atténuation des risques pour répondre à des conditions d’utilisation spécifiques nationales. Si, contrairement à l’Etat membre ayant examiné la demande, un Etat membre décide de refuser l’autorisation d’un produit phytopharmaceutique sur son territoire, il doit motiver son refus auprès de la société et de la Commission européenne.
→ L’autorisation est renouvelée sur demande de son titulaire, pour autant que les conditions permettant l’autorisation soient toujours remplies.
→ L’autorisation est modifiée ou retirée, selon les articles 44 et 45 du RCE n° 1107/2009, à la demande motivée du titulaire ou lorsque, le cas échéant :
- les exigences nécessaires à l’obtention de l’AMM présentées au IV.1 ne sont plus respectées ;
- des informations fausses ou trompeuses ont été fournies ;
- une condition figurant dans l’autorisation n’est pas remplie ;
- compte tenu de l’évolution des connaissances scientifiques et techniques, le mode d’utilisation et les quantités utilisées peuvent être modifiés ;
- le titulaire de l’autorisation ne respecte pas les obligations découlant du RCE n° 1107/2009 ;
Lorsqu’un Etat membre retire ou modifie une autorisation, il en informe immédiatement le titulaire, les autres Etats membres, la Commission et l’Autorité européenne de sécurité des aliments. Les autres Etats membres appartenant à la même zone retirent ou modifient l’autorisation en conséquence en tenant compte des paramètres nationaux et des mesures d’atténuation des risques. L’article 46 s’applique, le cas échéant.
L’article 46 du RCE n° 1107/2009 prévoit qu’en cas de retrait d’AMM, l’Etat membre peut accorder un délai de grâce pour l’élimination, le stockage, la mise sur le marché et l’utilisation des stocks existants. Ces délais de grâce sont limités et n’excèdent pas six mois pour la vente et la distribution, et un an supplémentaire (soit dix-huit mois au total) pour l’élimination, le stockage et l’utilisation des stocks existants de produits phytopharmaceutiques.
L’article 6 de l’ordonnance n° 2011-840 prévoit une période transitoire pour les demandes d’autorisation de mise sur le marché ainsi que les demandes de réexamen des AMM à la suite de l’approbation de la substance active, déposées avant le 14 juin 2011 à l’Anses.
Elles font l’objet d’une décision prise en application des dispositions du chapitre III du titre V du livre II du code rural et de la pêche maritime dans sa rédaction antérieure à la publication de l’ordonnance.
En France, tous les dossiers doivent être déposés auprès de l’Anses. Celle-ci dispose, selon les demandes (autorisation, modifications, renouvellement) d’un délai compris entre un mois (simple enregistrement de modification ne nécessitant pas d’évaluation) et dix mois pour examiner le dossier et rendre, le cas échéant, son avis.
La direction générale de l’alimentation, DGAL, du ministère chargé de l’agriculture, dispose d’un délai compris entre 5 jours et deux mois pour prendre sa décision.
Les décisions d’autorisation de mise sur le marché des produits sont publiées par voie électronique par l’Anses. A ce jour, l’Anses ne met en ligne que ses avis. Un site dédié aux décisions d’AMM est attendu.
(9) Directive 99/45/CE du Parlement européen et du Conseil du 31 mai 1999 concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à la classification, à l’emballage et à l’étiquetage des préparations dangereuses et le règlement (CE) n° 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l’étiquetage et à l’emballage des substances et des mélanges.
IV.4 Cas dérogatoires pour lesquels aucune autorisation de mise sur le marché n’est requise
Selon l’article 28 du RCE n° 1107/2009, aucune autorisation de mise sur le marché n’est requise dans les cas suivants :
- utilisation de produits contenant exclusivement une ou plusieurs substances de base ;
- mise sur le marché et utilisation de produits phytopharmaceutiques à des fins de recherche ou de développement ;
- production, stockage ou circulation d’un produit phytopharmaceutique destiné à être utilisé dans un autre État membre ;
- production, stockage ou circulation d’un produit phytopharmaceutique destiné à être utilisé dans un pays tiers ;
- mise sur le marché et utilisation de produits phytopharmaceutiques pour lesquels un permis de commerce parallèle a été accordé (voir IV.10).
IV.5 Cas particulier des produits phytopharmaceutiques « à faible risque »
Une procédure particulière est prévue à l’article 47 du RCE n° 1107/2009 pour les produits phytopharmaceutiques, dits « à faible risque ». Ces produits ne doivent contenir que des substances actives approuvées en tant que substance active à faible risque.
De plus, un produit « à faible risque » doit satisfaire aux conditions suivantes :
- les substances actives, phytoprotecteurs et synergistes à faible risque qu’il contient ont été approuvés ;
- il ne contient pas de substance préoccupante ;
- il est suffisamment efficace ;
- il ne provoque pas de souffrances ou de douleurs inacceptables chez les vertébrés à combattre ;
- ses coformulants ne figurent pas dans l’annexe III (coformulants inacceptables) ;
- la nature et la quantité de ses substances actives, phytoprotecteurs et synergistes et, le cas échéant, les impuretés et coformulants importants sur le plan toxicologique, écotoxicologique ou environnemental peuvent être déterminés à l’aide de méthodes appropriées ;
- les résidus résultant des utilisations autorisées peuvent être déterminés à l’aide de méthodes appropriées d’usage courant dans tous les Etats membres ;
- ses propriétés physico-chimiques ont été déterminées et jugées acceptables pour assurer une utilisation et un stockage adéquats du produit ;
- pour les végétaux ou produits végétaux devant, le cas échéant, être utilisés comme cultures fourragères ou vivrières, les limites maximales de résidus applicables aux produits agricoles concernés par l’utilisation visée dans l’autorisation ont été établies ou modifiées conformément au règlement (CE) n° 396/2005.
L’Anses a 90 jours pour rendre son avis. La DGAL a un mois pour prendre sa décision.
IV.6 Cas particulier français des préparations naturelles peu préoccupantes
L’article D.253-22 du code rural et de la pêche maritime définit les « préparations naturelles peu préoccupantes à usage phytopharmaceutique » comme tout produit qui est composé exclusivement d’une ou plusieurs substances de base ou d’une ou plusieurs substances active à faible risque. Dans ce dernier cas, il s’agit d’un produit phytopharmaceutique à faible risque (voir IV.5) soumis à autorisation de mise sur le marché.
En revanche, une préparation naturelle peu préoccupante à usage phytopharmaceutique composée exclusivement d’une ou plusieurs substances de base doit :
- pouvoir être obtenue par un procédé accessible à tout utilisateur final ;
- et être composée d'une ou plusieurs substances non traitées, ou traitées uniquement par des moyens manuels, mécaniques ou gravitationnels, par dissolution dans l'eau, par flottation, par extraction par l'eau, par distillation à la vapeur ou par chauffage uniquement pour éliminer l'eau ;
Un « procédé accessible à tout utilisateur final » est un procédé pour lequel l'utilisateur final est capable de réaliser toutes les étapes de la préparation. Néanmoins la matière première peut avoir été acquise auprès d'entreprises extérieures lorsque celles-ci sont seules capables de la fournir et si ces dernières ne réalisent pas elles-mêmes la préparation.
Il est rappelé que, conformément à l’article 28 du RCE n° 1107/2009 (voir IV.4), l’utilisation d’une préparation naturelle peu préoccupante à usage phytopharmaceutique composée exclusivement d’une ou plusieurs substances de base n’est pas soumise à autorisation de mise sur le marché (voir IV.4).
Par ailleurs l’article 6 de l’ordonnance n° 2011-840 prévoit que la mise sur le marché des préparations naturelles peu préoccupantes bénéficiant d’une autorisation de mise sur le marché délivrée conformément aux dispositions prises pour l’application du IV de l’article L. 253-1 du code rural et de la pêche maritime dans sa version antérieure à la date de publication de la présente ordonnance est autorisée jusqu’à la révision de l’autorisation suite à l’inscription de la ou des substances actives qu’elles contiennent en tant que substance, de base ou non, sur la liste positive communautaire des substances actives (RCE n°540/2011).
A défaut de demande d’approbation des substances concernées déposée avant le 31 décembre 2011, les autorisations de mise sur le marché des préparations naturelles peu préoccupantes seront considérées comme échues à cette date.
A ce jour, seules les préparations naturelles peu préoccupantes à base de purin d’ortie ont été autorisées en France par l’arrêté du 18 avril 2011 (voir note d’information n° 2011-46).
IV.7 Evaluation comparative des produits phytopharmaceutiques contenant une substance dont on envisage la substitution
L’article 50 du RCE n°1107/2009 prévoit que les Etats membres réalisent une évaluation comparative lors de l’examen de toute demande d’autorisation relative à un produit phytopharmaceutique contenant une substance active approuvée en tant que substance dont on envisage la substitution.
Les Etats membres n’autorisent pas ou limitent l’utilisation d’un produit phytopharmaceutique pour une culture donnée, qui contient une substance dont on envisage la substitution lorsqu’il ressort de l’évaluation comparative mettant en balance les risques et les bénéfices :
a) qu’il existe déjà, pour les utilisations précisées dans la demande, un produit phytopharmaceutique autorisé ou une méthode non chimique de prévention ou de lutte qui est sensiblement plus sûr pour la santé humaine ou animale ou l’environnement ;
b) et que la substitution par des produits phytopharmaceutiques ou des méthodes non chimiques de prévention ou de lutte visés au point a) ne présente pas d’inconvénients économiques ou pratiques majeurs ;
c) et que la diversité chimique des substances actives, le cas échéant, ou les méthodes et pratiques de gestion des cultures et de prévention des ennemis des cultures sont de nature à réduire autant que possible l’apparition d’une résistance dans l’organisme cible ;
d) et que les conséquences sur les autorisations pour des utilisations mineures (10) sont prises en compte.
Les principes de l’évaluation comparative sont énoncés à l’annexe IV du RCE n° 1107/2009.
(10) Utilisation mineure : utilisation d’un produit phytopharmaceutique, dans un Etat membre particulier, sur les végétaux ou produits végétaux qui ne sont pas largement cultivés dans cet Etat membre ou qui sont largement cultivés, pour répondre à un besoin exceptionnel en matière de protection des végétaux.
IV.8 Filiations entre les produits identiques autorisés en France
Les articles D.253-8 et D.253-9 du code rural et de la pêche maritime définissent désormais les différentes relations d’identités possibles entre des produits mis sur le marché français.
Une autorisation de mise sur le marché d’un produit phytopharmaceutique est délivrée pour l’une des deux gammes d’usages suivantes : la gamme d’usages professionnels et la gamme d’usages amateurs.
Chaque gamme comprend une liste d’usages spécifiques repris dans le catalogue des usages.
Le produit dit « de référence » est le produit qui a fait l’objet de la première autorisation de mise sur le marché en France dans une gamme. Ce même produit (formulation strictement identique dans toutes ses composantes : substances actives, coformulants) pourra être mis sur le marché :
- dans une autre gamme : il s’agit d’un produit dit « de seconde gamme » : la demande de « seconde gamme » est introduite par le titulaire de l’AMM du produit de référence.
- par une autre société, distincte du titulaire de l’AMM de référence : il s’agit d’un produit dit « de revente » ;
La demande concernant ce produit a été introduite par un demandeur distinct du titulaire de l’AMM après accord de ce dernier. Elle peut viser une liste d’usages identique ou différente de celle du produit de référence.
- sous une autre dénomination commerciale : il s’agit d’un produit dit « second nom commercial »
La demande concernant ce produit a été introduite par le titulaire de l’AMM du produit de référence. Elle vise à attribuer une nouvelle dénomination commerciale au produit, tout en restant dans la même gamme. La nouvelle dénomination commerciale couvre les mêmes usages que le produit de référence dans des conditions d’utilisation strictement identiques.
Les modifications intervenues sur les décisions d’autorisation de mise sur le marché concernant les produits de référence s’appliqueront aux « seconds noms commerciaux », aux « produits de seconde gamme » et aux « produits de revente » lorsque ces modifications concernent des mesures de gestion des risques (en vue de les atténuer) ou lorsqu’elles seront prises pour des motifs de santé publique ou de protection de l’environnement, dès lors que l’usage est couvert par le produit de référence. Cela est le cas par exemple lors d’une modification de dose d’emploi ou de délai avant récolte. Ces modifications sont publiées par voie électronique. Ainsi, a contrario, l’ajout d’un usage pour le produit de référence, à la demande du titulaire de l’AMM ne se répercute pas automatiquement sur les produits « second nom commercial », « seconde gamme » ou « revente ». Il faut une demande expresse des titulaires des AMM de ces produits.
L’article D.253-9 définit le produit phytopharmaceutique générique comme un produit ayant la même composition qualitative et quantitative en substance active et le même type de formulation qu’un produit de rattachement. Les effets devront être comparables à ceux du produit de rattachement.
Enfin, la mise en conformité des autorisations de mise sur le marché des produits dans ces nouvelles gammes d’usages et avec ces nouvelles définitions est échelonnée dans le temps. L’article 4 du décret n°2012-755 prévoit une mise en conformité au moment du renouvellement de l’autorisation de mise sur le marché du produit et au plus tard le 31 décembre 2016.
IV.9 Circulation des produits d’un Etat membre à l’autre : la reconnaissance mutuelle
Le principe de reconnaissance mutuelle est l’un des moyens de garantir la libre circulation des marchandises au sein de l’Union européenne.
Pour éviter les doubles emplois, réduire la charge administrative pesant sur l’industrie et les Etats membres et prévoir une mise à disposition plus harmonisée des produits phytopharmaceutiques, les autorisations accordées par un État membre doivent être acceptées par les autres États membres lorsque les conditions agricoles, phytosanitaires et environnementales (y compris climatiques) sont comparables.
C’est la raison pour laquelle l’Union européenne a été divisée en trois zones présentant des conditions comparables. Les trois zones sont définies à l’annexe 1 du règlement (CE) n° 1107/2009 :
→ Zone A – Nord : Danemark, Estonie, Lettonie, Lituanie, Finlande, Suède.
→ Zone B – Centre : Belgique, République tchèque, Allemagne, Irlande, Luxembourg, Hongrie, Pays-Bas, Autriche, Pologne, Roumanie, Slovénie, Slovaquie, Royaume-Uni.
→ Zone C – Sud : Bulgarie, Grèce, Espagne, France, Italie, Chypre, Malte, Portugal.
Dans le cas particulier des produits utilisés en serres, des produits de traitement après récolte, de traitement de semences ou de traitement des locaux, la distinction en trois zones n’a pas été reprise.
Les conditions agricoles, phytosanitaires et environnementales ont été jugées comparables pour les trois zones.
La reconnaissance mutuelle peut donc s’opérer :
- au sein d’une même zone, si l’autorisation a déjà été accordée par un Etat membre (Etat membre de référence) ;
- entre deux zones, si l’autorisation a déjà été accordée par un Etat membre d’une zone différente. L’autorisation accordée, sous reconnaissance mutuelle interzonale, ne pourra servir de référence pour une reconnaissance mutuelle intrazonale.
La demande de reconnaissance mutuelle est déposée par le titulaire de l’AMM du produit octroyée dans l’Etat membre de référence (situation différente de celle prévue par la procédure de permis de commerce parallèle, voir IV.10).
Le produit autorisé en procédure de reconnaissance mutuelle l’est pour les mêmes conditions que le produit de référence, sauf s’il est jugé nécessaire par l’Etat membre destinataire d’ajouter des mesures d’atténuation des risques dûment motivées.
Les délais d’examen des demandes sont réduits à 120 jours (90 jours pour l’Anses et un mois pour la DGAL).
IV.10 Circulation des produits d’un Etat membre à l’autre : le permis de commerce parallèle
Selon l’article 52 du RCE n° 1107/2009, un produit phytopharmaceutique qui est autorisé dans un Etat membre (Etat membre d’origine) peut, sous réserve de l’octroi d’un permis de commerce parallèle, être introduit, mis sur le marché ou utilisé dans un autre État membre (État membre d’introduction) si ce dernier établit que la composition du produit phytopharmaceutique est identique à celle d’un produit phytopharmaceutique déjà autorisé sur son territoire (produit de référence).
La demande est adressée à l’autorité compétente de l’Etat membre d’introduction.
A la différence de la procédure de reconnaissance mutuelle, le demandeur qui deviendra le titulaire du permis de commerce parallèle, n’est pas le détenteur de l’AMM du produit de référence. Il n’a donc accès à aucun dossier d’évaluation. C’est l’Etat membre d’introduction qui sollicite le dossier auprès de l’Etat membre d’origine.
Cette procédure s’applique quelles que soient les zones des Etats membres d’origine et d’introduction.
Le permis de commerce parallèle est accordé selon une procédure simplifiée dans un délai de 60 jours (45 jours pour l’Anses et 15 jours pour la DGAL)
Les produits phytopharmaceutiques sont réputés identiques aux produits de référence :
a) s’ils ont été fabriqués par la même société ou par une société associée ou sont fabriqués sous licence selon le même procédé de fabrication ;
b) et s’ils sont identiques pour ce qui est de la spécification, de la teneur et du type de formulation aux substances actives, phytoprotecteurs et synergistes et du type de formulation ;
c) et s’ils sont identiques ou équivalents en ce qui concerne les coformulants présents et la dimension, le matériau ou la forme de l’emballage, pour ce qui est de l’impact négatif potentiel sur la sécurité du produit en ce qui concerne la santé humaine ou animale ou l’environnement.
Un produit phytopharmaceutique pour lequel un permis de commerce parallèle a été délivré est mis sur le marché et utilisé uniquement conformément aux dispositions de l’autorisation du produit de référence.
Le permis de commerce parallèle est valable pendant la durée de l’autorisation du produit de référence. Si le titulaire de l’autorisation du produit de référence demande le retrait de l’autorisation, et si les exigences de l’article 29 sont toujours remplies, la validité du permis de commerce parallèle expire à la date à laquelle l’autorisation du produit de référence arriverait normalement à échéance.
Un permis de commerce parallèle peut être retiré si l’autorisation du produit phytopharmaceutique introduit est retirée dans l’État membre d’origine pour des raisons de sécurité ou d’efficacité.
L’article R.253-26 du code rural et de la pêche maritime prévoit la mise à disposition du public par voie électronique de la liste régulièrement actualisée des produits dont l'introduction est permise sur le territoire national suite à la délivrance d’un permis de commerce parallèle. Cette liste mentionnera pour chaque produit sous permis de commerce parallèle : l'Etat membre d'origine, le produit de référence ainsi que les mentions d’étiquetage obligatoires en langue française du produit de référence.
La liste ainsi publiée vaudra permis de commerce parallèle pour un usage personnel pour chacun des produits qui y sont listés (voir IX).
Selon l’article R.253-29 du code rural et de la pêche maritime, le ministre chargé de l’agriculture pourra retirer ou modifier le permis de commerce parallèle :
- en cas de constatation de non-conformité sur un lot de produits lorsque cette non-conformité consiste à substituer volontairement un autre produit au produit d’origine pour lequel le permis a été délivré,
- en cas de constatation de non-conformité d’un lot de produits laissant supposer que tout ou partie des produits mis sur le marché ne remplissent pas les conditions pour lesquelles le permis de commerce parallèle a été délivré et sont susceptibles de présenter un risque pour la santé publique et l’environnement.
Enfin, l’article 4 (V) du décret n° 2012-755 du 9 mai 2012 prévoit que les autorisations de mise sur le marché à titre d’introduction parallèle délivrées avant le 1er juillet 2012 (entrée en vigueur du décret précité) valent permis de commerce parallèle.
IV.11 Règles particulières pour les adjuvants
L’article 58 du RCE n° 1107/2009 prévoit que la mise sur le marché et l’utilisation des adjuvants sont soumises à autorisation dans chaque Etat membre concerné.
Un règlement spécifique définira les modalités d’autorisation ainsi que les exigences en matière de données et d’évaluation.
Dans l’attente de la publication de ce futur règlement spécifique, l’article 6 de l’ordonnance n°2011- 840 prévoit que la mise sur le marché et l’utilisation des adjuvants sont soumises aux dispositions du chapitre III du titre V du livre II du code rural et de la pêche maritime dans leur rédaction issue de la présente ordonnance, c’est-à-dire aux mêmes dispositions que celles prévues dorénavant pour les produits phytopharmaceutiques (autorisation de mise sur le marché).
V. Exigences relatives à l’emballage, l’étiquetage et la publicité des produits phytopharmaceutiques
V.1 L’emballage
L’article 64 du RCE n°1107/2009 prévoit que les produits phytopharmaceutiques et les adjuvants susceptibles d’être pris à tort pour des denrées alimentaires, des boissons ou des aliments pour animaux doivent être emballés de façon à réduire autant que possible la probabilité de telles méprises.
En outre, les produits phytopharmaceutiques et les adjuvants accessibles au grand public et susceptibles d’être pris à tort pour des denrées alimentaires, des boissons ou des aliments pour animaux doivent contenir des composants propres à dissuader ou empêcher leur consommation.
L’article 9 de la directive 1999/45/CE (11) transposé à l’article 22 de l’arrêté du 9 novembre 2004 (12) s’applique également aux produits phytopharmaceutiques et aux adjuvants non régis par ladite directive. Ainsi, les emballages de tous les produits phytopharmaceutiques et adjuvants, quel que soit leur classement de danger, doivent être conçus et réalisés de manière à empêcher toute déperdition du contenu (sauf dispositif de sécurité spécial prescrit). Les matières dont sont constitués les emballages et les fermetures ne doivent pas être susceptibles d’être attaquées par le contenu, ni de former avec ce dernier des composés dangereux. Toutes les parties des emballages et des fermetures doivent être solides et résistantes de manière à exclure tout relâchement et à répondre en toute sécurité aux tensions et efforts normaux de manutention.
En complément de ces exigences, l’article R.253-43 du code rural et de la pêche maritime ajoute que les produits phytopharmaceutiques et les adjuvants doivent être mis sur le marché et conservés dans leur contenant et emballage d'origine jusqu'au moment de leur utilisation. Les emballages utilisés pour les besoins des opérations de manutention devront présenter les mêmes garanties que celles qui étaient assurées par l'emballage d'origine. Il demeure une exception pour les produits bénéficiant d’un permis de commerce parallèle. A la demande du titulaire du permis de commerce parallèle, le ministre chargé de l’agriculture peut autoriser le reconditionnement sous réserve du respect des conditions suivantes :
- le reconditionnement est nécessaire pour accéder au marché national, en raison de ses contraintes spécifiques liées à l’emballage ou au contenant du produit ;
- l’intégrité et la traçabilité du produit introduit sont garanties ;
- le titulaire de l’autorisation de mise sur le marché du produit introduit a été préalablement informé du reconditionnement envisagé.
(11) Directive 1999/45/CE du Parlement européen et du Conseil du 31 mai 1999 concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à la classification, à l’emballage et à l’étiquetage des préparations dangereuses.
(12)Arrêté du 9 novembre 2004 définissant les critères de classification et les conditions d’étiquetage et d’emballage des préparations dangereuses et transposant la directive 1999/45/CE.
V.2 L’étiquetage
V.2.1 L’étiquetage de danger
Selon l’article 65 du RCE n° 1107/2009, l’étiquetage des produits phytopharmaceutiques inclut les exigences en matière de classification, d’étiquetage et d’emballage de la directive 1999/45/CE précitée transposée par l’arrêté du 9 novembre 2004.
Ainsi, l’étiquetage de danger d’un produit phytopharmaceutique découle de la classification de danger du produit établie sur la base des règles énoncées pour les préparations ou mélanges dangereux (règles fondées sur des résultats d’essais ou des calculs). Il se compose notamment de pictogrammes, d’indications de danger, de phrases de risques et de conseils de prudence.
Par ailleurs, l’article 24 (III) de l'arrêté du 9 novembre 2004 précité exige, pour tout produit phytopharmaceutique, l’apposition de la mention dénommée Sp1 : « respectez les instructions d’utilisation pour éviter les risques pour l’homme et l’environnement ». (13)
En cas d’évolution de la réglementation sur les préparations ou mélanges dangereux (règlement d’adaptation au progrès technique et scientifique dit ATP), la classification et l’étiquetage de danger sont susceptibles d’évoluer. Les délais de mise en conformité des étiquetages sont alors ceux fixés par ces règlements ATP. Il y a généralement un délai de 12 à 18 mois entre la publication du règlement ATP et son entrée en application. Les étiquetages des produits mis sur le marché à compter de l’entrée en application du règlement ATP doivent être conformes à ce règlement ATP.
Par ailleurs, l’article R.253-42 (I) du code rural et de la pêche maritime exige la notification au ministère chargé de l’agriculture des modifications de classification et d’étiquetage par le titulaire de l’autorisation de mise sur le marché ou du permis de commerce parallèle au plus tard deux mois avant la date d’entrée en application du règlement ATP. Le ministre chargé de l’agriculture publie le nouveau classement du produit par voie électronique dans un délai maximum de deux mois à compter de la date de notification du changement de classement.
Enfin, l’article R.253-42 (I) du code rural et de la pêche maritime détermine des délais maximum d’écoulement des produits dont la première mise sur le marché est antérieure à l’entrée en application du règlement ATP. Les produits peuvent être commercialisés pendant une période de six mois suivant la date d’application du règlement ATP et ils peuvent être utilisés pendant dix-huit mois à compter de la date d’application du règlement ATP.
(13) Il faut signaler qu’au 14 juin 2015, l’arrêté du 9 novembre 2004 sera abrogé et remplacé par le règlement 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) n° 1907/2006. Ainsi, l’obligation de l’article 24 (III) sera remplacée par celle prévue à l’annexe II point 4 de ce règlement. Il s’agira d’apposer la mention dite EUH401 sur tous les produits phytopharmaceutiques. Le libellé sera le même : « Respectez les instructions d'utilisation pour éviter les risques pour la santé humaine et l'environnement ».
V.2.2 L’étiquetage phytopharmaceutique
En application de l’article 65 du RCE n°1107/2009, les mentions d’étiquetage des produits phytopharmaceutiques sont celles prévues par le règlement (UE) n°547/2011. L’annexe de ce règlement liste 21 mentions d’étiquetage auxquelles s’ajoutent, le cas échéant, des phrases types sur les risques particuliers encourus et les précautions à prendre qui complètent l’étiquetage de danger, selon des critères précisés également en annexe du règlement (UE) n° 547/2011 (phrases dites RSh, Spe, Spr, Spo).
Cependant, l’article 80 du RCE n° 1107/2009 repris également par l’article 6 de l’ordonnance n° 2011-840, permet que les produits phytopharmaceutiques étiquetés conformément à l’article 16 de la directive 91/414/CEE, transposé par l’article 34 de l’arrêté du 6 septembre 1994 (14) peuvent continuer à être mis sur le marché jusqu’au 14 juin 2015.
Ainsi, d’ici le 14 juin 2015, les responsables de la mise sur le marché des produits phytopharmaceutiques et des adjuvants ont le choix entre appliquer les mentions prévues par le règlement (UE) n°547/2011 ou celles prévues par l’article 34 de l’arrêté du 6 septembre 1994. Une fois cette date passée, tous les produits sur le marché devront être étiquetés conformément au règlement (UE) n°547/2011.
(14) Arrêté du 6 septembre 1994 modifié portant application du décret n°94-359 du 5 mai 1994 relatif au contrôle de la mise sur le marché des produits phytopharmaceutiques
Le tableau comparatif ci-après permet de signaler les quelques différences entre l’étiquetage prévu par le règlement (UE) n° 547/2011 et celui prévu par l’article 34 de l’arrêté du 6 septembre 1994 :
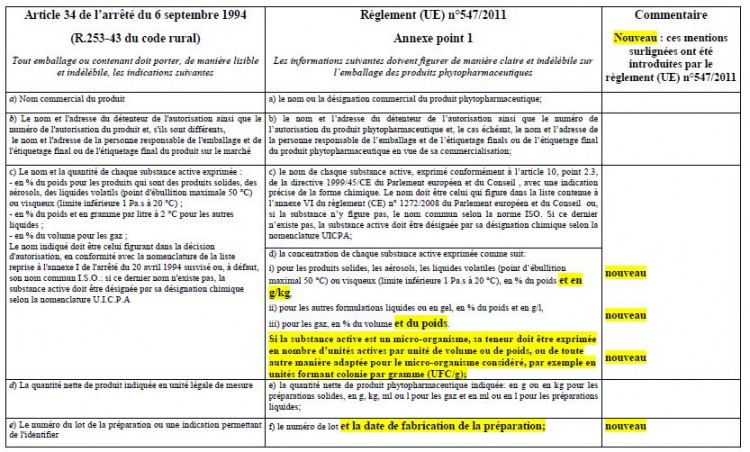
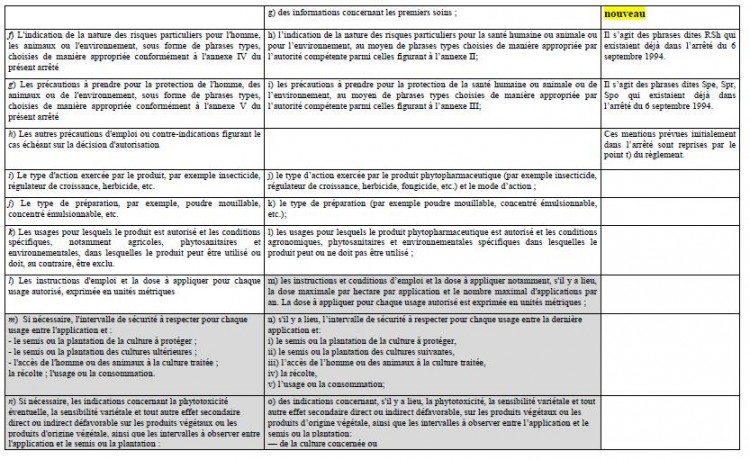
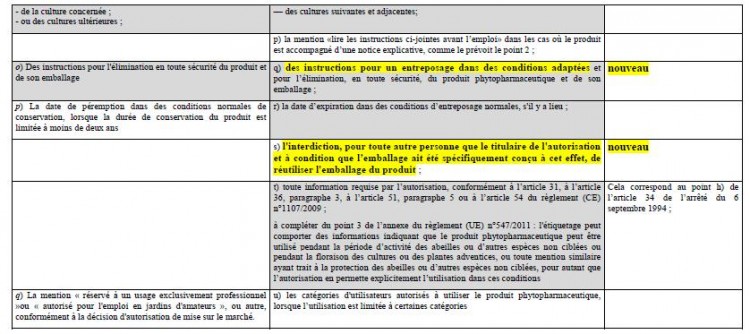
En grisé sur le tableau : la liste des mentions pouvant apparaître sur une notice séparée accompagnant l’emballage, soit en application de l’article 35 de l’arrêté du 6 septembre 1994, soit en application du point 2 de l’annexe I du règlement (UE) n° 547/2011. Dans les deux cas, l'emballage ou le contenant doivent porter la phrase: « Lire les instructions ci-jointes avant l'emploi ».
En outre, il doit être signalé deux dérogations d’étiquetage qui n’ont pas été reprises dans le règlement (UE) n° 547/2011 :
→ Cas particulier des emballages contenant des petits conditionnements prêts à l’emploi,
l’article 36 de l’arrêté du 6 septembre 1994 prévoit que ces derniers peuvent ne comporter que les mentions suivantes :
a) le nom commercial ou désignation du produit ;
b) nom et adresse du demandeur responsable de la mise sur le marché ;
c) symboles et indications de danger le cas échéant ;
d) le nom des substances actives et des substances dangereuses prévues par la réglementation en vigueur.
→ Cas particulier des emballages contenant des sachets hydrosolubles, l’article 37 de l’arrêté du 6 septembre 1994 prévoit que ces derniers doivent porter au moins les indications suivantes :
a) le nom commercial ou désignation du produit ;
b) « sachet hydrosoluble ou soluble : à conserver dans l'emballage d'origine à l'abri de l'humidité ; se référer à conditions et précautions d'emploi mentionnées sur l'emballage ». Dans ce cas, l'emballage ou le contenant doit porter des indications appropriées sur le mode d'emploi des sachets hydrosolubles.
En revanche, le règlement (UE) n° 547/2011 précise le cas des étiquetages et emballages des produits phytopharmaceutiques destinés à être utilisés pour des expériences ou des essais à des fins de recherche ou de développement (point 5 de l’annexe). Ces derniers doivent uniquement être conformes au point 1, sous b), c), d), j) et k). L’étiquetage comporte les informations requises par le permis délivré pour effectuer des essais, prévu à l’article 54, paragraphe 1, du règlement (CE) n° 1107/2009, et la mention « Produit destiné à un usage expérimental dont la spécification est incomplète. A manipuler avec une extrême prudence ».
En outre, il est précisé au point 3 de l’annexe que l’étiquetage d’un produit phytopharmaceutique ne peut comporter de mentions telles que « non toxique », « ne nuit pas à la santé » ou toute indication similaire.
Enfin, l’article R.253-42 (II) du code rural et de la pêche maritime prévoit des délais maximum de mise à jour et d’écoulement des produits en cas de modification de l’autorisation de mise sur le marché entraînant la modification de l’étiquetage (changement de dose, de délai avant récolte…) :
Le titulaire de l’autorisation de mise sur le marché met sur le marché des produits étiquetés conformément à l’autorisation dans un délai de six mois à compter de la notification de l’autorisation de mise sur le marché de modification et met à jour les étiquettes des produits commercialisés dans ce même délai. Dans les cas où la modification de la décision d’autorisation de mise sur le marché consiste en un élargissement des usages du produit ou en un allégement de ses précautions d’utilisation, ce délai est porté à un an.
Les stocks de produits dont la première mise sur le marché français est antérieure à la date limite de mise à jour des étiquettes des produits, peuvent être utilisés pendant une période de douze mois à compter de cette date limite de mise à jour (soit dix-huit mois à compter de la notification de l’autorisation de mise sur le marché de modification).
Ces délais de mise à jour des étiquettes et d’écoulement des stocks ne s’appliquent pas dans les cas où la décision de modification de l’autorisation de mise sur le marché ou un arrêté du ministre chargé de l’agriculture prévoient des délais différents.
V.3 La publicité
L’article 66 du règlement RCE n° 1107/2009 prévoit que seuls les produits phytopharmaceutiques autorisés peuvent faire l’objet de publicité.
Toute publicité pour un produit phytopharmaceutique doit être accompagnée des phrases « Utilisez les produits phytopharmaceutiques avec précaution. Avant toute utilisation, lisez l’étiquette et les informations concernant le produit ». Ces phrases sont aisément lisibles et doivent se distinguer clairement de l’ensemble de la publicité. Les mots « produits phytopharmaceutiques » peuvent être remplacés par une description plus précise du type de produit tel que fongicide, insecticide ou herbicide.
La publicité ne peut pas comporter d’informations potentiellement trompeuses, sous forme de textes ou d’illustrations, sur les risques éventuels pour la santé humaine ou animale ou l’environnement, telles que les termes « à faible risque », « non toxique » ou « sans danger ». Les termes « autorisé comme produit phytopharmaceutique à faible risque conformément au règlement (CE) n° 1107/2009 » ne sont autorisés dans la publicité que dans le cas des produits phytopharmaceutiques à faible risque.
Ils ne peuvent être utilisés comme allégation sur l’étiquette du produit phytopharmaceutique.
Toutes les allégations publicitaires doivent se justifier sur le plan technique.
Enfin, les publicités ne contiennent aucune représentation visuelle de pratiques potentiellement dangereuses telles que le mélange ou l’application sans vêtements de protection suffisants, l’utilisation à proximité des denrées alimentaires, ou l’utilisation par des enfants ou à proximité de ceux-ci.
Le matériel publicitaire ou promotionnel attire l’attention sur les phrases et les symboles de mise en garde appropriés figurant sur l’étiquetage. Sous réserve de l’appréciation souveraine des tribunaux, il s’agira d’apposer sur tout prospectus, catalogue, site internet (etc.) les pictogrammes, phrases de risques et conseils de prudence issus de l’étiquetage de danger des produits phytopharmaceutiques.
Par ailleurs, l’article L.253-5 du code rural interdit la publicité commerciale destinée au grand public, télévisée, radiodiffusée et par voie d’affichage extérieur en dehors des points de distribution pour les produits phytopharmaceutiques et les adjuvants. Cela ne vise pas les sites internet d’information ou de vente, ni les catalogues promotionnels.
Il est prévu qu’un arrêté (non encore paru à ce jour) fixera les conditions de présentation des bonnes pratiques d’utilisation et d’application de ces produits dans les insertions publicitaires, en tenant compte des différences entre produits destinés aux professionnels et produits destinés aux particuliers.
Ces insertions publicitaires mettront en avant les principes de la lutte intégrée et de bonnes pratiques dans l’usage et l’application des produits.
VI. Cas particulier des produits phytopharmaceutiques destinés au grand public
VI.1 Textes réglementaires spécifiques concernant les produits phytopharmaceutiques destinés au grand public
La mise sur le marché des produits phytopharmaceutiques destinés au grand public est réglementée par :
→ les articles D.253-8 et R.253-41 du code rural et de la pêche maritime introduits par le décret n° 2012-755 du 9 mai 2012 ;
→ les articles R.254-20, R.254-21, R.254-22 et R.254-30 du code rural et de la pêche maritime introduits par le décret n° 2011-1325 du 18 octobre 2011 (voir VI.2) ;
→ l’arrêté du 30 décembre 2010 interdisant l'emploi de certains produits phytopharmaceutiques par des utilisateurs non professionnels (NOR : AGRG 1030490A - voir VI.2).
→ l’arrêté du 30 décembre 2010 relatif aux conditions d'emballage des produits phytopharmaceutiques pouvant être employés par des utilisateurs non professionnels (NOR : AGRG 1030485A - voir VI.3) ;
VI.2 Conditions d’autorisation des produits phytopharmaceutiques destinés au grand public
Les produits phytopharmaceutiques destinés à être utilisés par le grand public sont autorisés dans la gamme d’usages « amateur ». Les conditions d’attribution de la mention sont énoncées à l’article D.253-8 du code rural et de la pêche maritime. Seuls peuvent être autorisés pour la gamme d’usages « amateur » les produits :
- dont la formulation et le mode d’application sont de nature à garantir un risque d’exposition limité pour l’utilisateur. l’arrêté du 30 décembre 2010 (NOR : AGRG 1030490A) précise les catégories de produits ne répondant pas à ce critère ;
- et dont l’emballage et l’étiquette proposés, outre qu’ils sont conformes aux exigences réglementaires relatives aux conditions d’étiquetage, répondent aux conditions fixées par l’arrêté du 30 décembre 2010 (NOR : AGRG 1030485A).
En application du point 4 d) de l’article 31 du RCE n° 1107/2009, l’autorisation de mise sur le marché précise la catégorie d’utilisateurs (professionnels / non-professionnels) pour laquelle est destiné le produit.
En application de l’article 1er de l’arrêté du 30 décembre 2010 interdisant l'emploi de certains produits phytopharmaceutiques par des utilisateurs non professionnels ((NOR : AGRG 1030490A), les produits phytopharmaceutiques suivants ne peuvent être autorisés pour le grand public :
1. Les produits classés dans les catégories explosifs, très toxiques (T +), toxiques (T), cancérogènes, mutagènes ou encore toxiques ou nocifs pour la reproduction ou le développement, correspondant aux phrases de risque :
R. 40, R. 60, R. 61, R. 62, R. 63, R. 68, R. 45, R. 46, R. 49 (classification selon l’arrêté du 9 novembre 2004)
ou
H200, H201, H202, H203, H204, H205, H300, H301, H310, H311, H330, H331, H370, H372, H350 et H350i, H340, H360F, H360D, H360FD, H360Fd H360Df, H351, H341, H361f, H361d, H361fd (classification
selon le règlement (CE) n°1272/2008) ;
2. Les produits contenant les substances actives suivantes :
a) Les substances répondant aux critères de classification comme substances cancérogènes, de catégorie 1A ou 1B, conformément au règlement (CE) n° 1272/2008, correspondant aux mentions de danger suivantes : H350 et H350i ;
b) Les substances répondant aux critères de classification comme substances mutagènes, de catégorie 1A ou 1B, conformément au règlement (CE) n° 1272/2008, correspondant à la mention de danger suivante : H340 ;
c) Les substances répondant aux critères de classification comme substances toxiques pour la reproduction, de catégorie 1A ou 1B, conformément au règlement (CE) n° 1272/2008, correspondant aux mentions de danger suivantes : H360 F, H360D, H360 FD, H360 Fd H360Df ;
d) Les substances qui sont persistantes, bioaccumulables et toxiques, conformément aux critères énoncés à l’annexe XIII du règlement (CE) n°1907/2006 du Parlement européen et du Conseil du 18 décembre 2006 ;
e)Les substances qui sont très persistantes et très bioaccumulables, conformément aux critères énoncés à l’annexe XIII du règlement (CE) n° 1907/2006, ou si la classification de ces substances comporte les phrases de risque R. 45, R. 46, R. 49, R. 60 ou R. 61 (classification selon l’arrêté du 20 avril 1994) ;
3. Les produits destinés au traitement des cultures vivrières si aucune limite maximale de résidus n’a été préalablement définie pour les substances actives qu’ils contiennent et les cultures visées par le traitement ;
4. Les produits de lutte contre les ragondins, les campagnols, les rats musqués, les mulots et taupicides présentés sous forme de concentrés liquides pour préparation d’appâts et de poudres de piste, ou formulés avec des
miettes de pain comme support d’appâts ;
5. Les produits de lutte contre les ragondins, les campagnols, les rats musqués, les mulots et taupicides présentés sous forme de concentrés liquides pour préparation d’appâts et de poudres de piste, ou formulés avec des miettes de pain comme support d’appâts.
En outre, s’agissant de produits phytopharmaceutiques de lutte contre les rongeurs ravageurs des cultures destinés au grand public, ces produits doivent contenir un agent d’amertume à raison, sauf études validées par l’Anses permettant d’abaisser ces teneurs :
- de 10 ppm pour les préparations à base de grains ;
- de 50 ppm pour les préparations à base de granulés et de pâtes molles ;
- de 100 ppm pour les préparations à base de blocs paraffinés.
VI.3 Emballage et étiquetage des produits phytopharmaceutiques destinés au grand public
L’article R.253-41 du code rural et de la pêche maritime prévoit l’obligation d’apposer visiblement la mention « emploi autorisé dans les jardins » pour les produits autorisés ou couverts par un permis de commerce parallèle (ou une mention équivalente dans le cas de produits en provenance d’un autre Etat membre de l’Union européenne).
L’article 1er de l’arrêté du 30 décembre 2010 relatif aux conditions d'emballage des produits phytopharmaceutiques destinés au grand public (NOR : AGRG 1030485A) prévoit que les emballages de ces produits doivent répondre aux conditions suivantes :
1. L’emballage ou l’étiquetage mentionne un seul nom commercial figurant sur la décision d’autorisation. Le nom commercial unique et le numéro d’autorisation sont clairement indiqués sans être séparés par d’autres indications sous la forme :
« Nom homologué : ........................................................... N° d’AMM : ............................................ » ;
2. L’emballage ou l’étiquetage porte de manière lisible et indélébile les usages pour lesquels le produit est autorisé et les conditions spécifiques, notamment agronomiques, phytosanitaires et environnementales, dans lesquelles le produit doit être utilisé ou, au contraire, ne doit pas l’être, tels que prévus par l’autorisation de mise sur le marché ;
3. La mention du ou des usages principaux revendiqués figure sur la même face que le nom homologué ;
4. Les doses d’emploi sont indiquées en g ou ml/l, en g ou ml/5 l, en g ou ml/m2 ou en g ou ml/10 m2 ou en toute unité de dose prévue par la décision d’autorisation de mise sur le marché du produit après avis de l’Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail ;
5. Le délai avant récolte fixé par l’autorisation de mise sur le marché est indiqué sur l’emballage. A défaut d’indication dans l’autorisation, le délai indiqué sur l’emballage ou l’étiquetage est supérieur à cinq jours ;
6. L’emballage ou l’étiquetage ne comporte aucune mention pouvant suggérer une utilisation professionnelle du produit ou donner une image exagérément sécurisante ou de nature à banaliser l’utilisation du produit, notamment les mentions « non dangereux », « non toxique », « biodégradable » ;
7. L’emballage ou l’étiquetage garantit des conditions d’expositions minimales pour l’utilisateur et l’environnement.
A l’exception des unidoses, l’emballage est notamment refermable de façon étanche ou garantissant la sécurité de l’utilisateur ;
8. Toute mention ou tout pictogramme relatifs aux préconisations, notamment aux périodes de traitement favorables et toutes indications complémentaires relatives aux doses, doivent, pour pouvoir figurer sur l’étiquette, avoir été préalablement validés par l’agence nationale chargée de la sécurité sanitaire de l’alimentation, de l’environnement t du travail ;
9. Tout conditionnement associant plusieurs produits de lutte contre les ragondins, les campagnols, les rats musqués, les mulots est interdit.
Aucune sanction pénale n’est prévue pour le non-respect de ces dispositions. Toutefois, des mesures de police administrative peuvent être entreprises sur des lots de produits non conformes en application de l’article L.218-5 du code de la consommation.
VI.4 Conditions de vente des produits phytopharmaceutiques destinés au grand public
L’article R.254-20 du code rural et de la pêche maritime prévoit que les distributeurs ne mettent en vente, vendent ou distribuent au grand public, c’est-à-dire des personnes qui ne sont pas des « utilisateurs professionnels », que des produits dont l’autorisation comporte la mention « emploi autorisé dans les jardins ».
La notion d’utilisateur professionnel est définie à l’article R.254-1 du code rural et de la pêche maritime, comme toute personne qui utilise les produits phytopharmaceutiques dans le cadre de son activité professionnelle à titre salarié, pour son propre compte ou dans le cadre d’un contrat d’entraide à titre gratuit.
Le fait de céder à titre onéreux ou gratuit à des utilisateurs non professionnels un produit phytopharmaceutique dont l’autorisation de mise sur le marché ne prévoit pas qu’il peut leur être destiné est une infraction sanctionnée à l’article R.254-30 par une peine d’amende (contravention 5ème classe).
En application de l’article R.254-21 du code rural et de la pêche maritime, les produits dont l’autorisation comporte la mention « emploi autorisé dans les jardins » sont présentés à des emplacements séparés physiquement des produits dont l’autorisation ne comporte pas cette mention.
Ces deux catégories de produits sont indiquées à l’aide d’une signalétique explicite. Le fait de ne pas respecter cette disposition est sanctionné à l’article R.254-30 du code rural et de la pêche maritime par une peine d’amende (contravention 4ème classe).
Enfin, il est ajouté à l’article R.254-22 du code rural et de la pêche maritime que les utilisateurs nonprofessionnels reçoivent des informations générales sur les risques pour la santé humaine et l’environnement de l’utilisation des produits phytopharmaceutiques, notamment sur les dangers, l’exposition, les conditions appropriées de stockage et les consignes à respecter pour la manipulation, l’application et l’élimination sans danger de ces produits ainsi que, le cas échéant, sur les solutions de substitution présentant un faible risque. Cette disposition découle de l’article 6 de la directive n° 2009/128/CE (voir I.2) qui précise que cette obligation incombe aux distributeurs. Cette obligation sera d’ailleurs reprise dans le référentiel de certification pour les distributeurs de produits phytopharmaceutiques destinés au grand public (voir IX).
VII. Cas particulier des produits phytopharmaceutiques destinés au traitement de semences
L’article 49 du RCE n° 1107/2009 encadre la mise sur le marché des semences traitées avec des produits phytopharmaceutiques.
1. Les Etats membres n’interdisent pas la mise sur le marché et l’utilisation de semences traitées à l’aide de produits phytopharmaceutiques autorisés pour cette utilisation dans un État membre au moins.
2. (…)
3. (…)
4. Sans préjudice d’autres dispositions de la législation communautaire concernant l’étiquetage des semences, l’étiquette et les documents accompagnant les semences traitées mentionnent le nom du produit phytopharmaceutique avec lequel les semences ont été traitées, le(s) nom(s) de la (des) substance(s) active(s) présente(s) dans le produit, les phrases types pour les précautions en matière de sécurité prévues dans la directive 1999/45/CE et les mesures d’atténuation des risques énoncées dans l’autorisation de ce produit le cas échéant.
Les emballages de semences traitées avec des produits phytopharmaceutiques commercialisées en France doivent donc porter la mention :
→ du nom autorisé du produit phytopharmaceutique avec lequel les semences ont été traitées ;
→ des noms des substances actives du produit phytopharmaceutique avec lequel les semences ont été traitées :
→ des phrases types pour les précautions en matière de sécurité prévues dans la directive 1999/45/CE c’est-à-dire les conseils de prudence prévus (phrases S) attribués au produit phytopharmaceutique, présents sur l’étiquette et la fiche de données de sécurité du produit phytopharmaceutique ;
→ des mesures d’atténuation des risques énoncées dans l’autorisation de mise sur le marché du produit. Il s’agit notamment des phrases Spe, Spo, Spr.
VIII. Les produits « frontières »
VIII.1 Les produits mixtes : matières fertilisantes et phytopharmaceutiques
Les produits mixtes regroupent les produits revendiquant des propriétés phytopharmaceutiques et fertilisantes.
Au sens de l’article L.255-1 du code rural et de la pêche maritime, les matières fertilisantes comprennent les engrais, les amendements et, d'une manière générale, tous les produits dont l'emploi est destiné à assurer ou à améliorer la nutrition des végétaux ainsi que les propriétés physiques, chimiques et biologiques des sols. Leur mise sur le marché est conditionnée au respect des articles L.255-1 à L.255-11 du code rural et de la pêche maritime qui prévoient notamment leur conformité :
- soit à une autorisation de mise sur le marché ;
- soit à un règlement communautaire ;
- soit à une norme rendue d’application obligatoire.
Ainsi la mise sur le marché et l’utilisation des produits mixtes sont soumises au respect simultané des conditions prévues par les deux réglementations phytopharmaceutiques et matières fertilisantes.
VIII.2 Les produits biocides
Les produits biocides sont définis à l’article 2 de la directive 98/8/CE du 16 février 1998 concernant la mise sur le marché des produits biocides, transposée à l’article L.522-1 du code de l’environnement.
Ce sont des préparations à base de substances actives chimiques ou biologiques destinées à détruire, repousser, rendre inoffensif les organismes nuisibles, à en prévenir l’action ou à les combattre de toute autre manière par une action chimique ou biologique. Les organismes nuisibles sont définis comme tout organisme dont la présence n’est pas souhaitée ou qui produit un effet nocif pour l’homme, ses activités ou les produits qu’il utilise ou produit, ou pour les animaux ou pour l’environnement.
Cette définition englobe actuellement 23 types de produits biocides (désinfectants, produits de protection, produits antiparasitaires…) listés à l’article R.522-9 du code de l’environnement et détaillés à l’annexe V de l’arrêté du 19 mai 2004 modifié relatif au contrôle de la mise sur le marché des substances actives biocides et à l’autorisation de mise sur le marché des produits biocides.
Certains produits, tels que les rodenticides, les avicides, les molluscicides, les insecticides, les acaricides, les répulsifs et les appâts sont susceptibles d’être destinés à un usage phytopharmaceutique (protection des cultures) et / ou un usage biocide (protection de l’homme, de ses activités, des animaux…). En effet, les mêmes organismes vivants peuvent être nuisibles à des plantes comme à l’homme ou les animaux. Dans ce cas, les moyens de lutte peuvent être de composition identique même si les conditions d’emploi varient selon qu’il s’agit de protéger des cultures ou protéger l’homme, ses activités, les animaux ou l’environnement.
Or des réglementations distinctes ont été mises en place pour encadrer les produits phytopharmaceutiques et les produits biocides. L’article 1er de la directive 98/8/CE précitée exclut explicitement de son champ d’application les produits phytopharmaceutiques. Ainsi, dans l’état actuel de la réglementation européenne, de tels produits ne peuvent être commercialisés pour ces deux usages phytopharmaceutiques et biocides simultanément. Concrètement, un opérateur souhaitant mettre sur le marché un produit destiné à ces deux usages, devra obtenir une autorisation de mise sur le marché pour chacun de ces usages et différencier clairement les deux produits. Ainsi l’étiquetage sera différent bien que la composition soit la même.
Le classement d’un produit donné dans le domaine biocide ou phytopharmaceutique s’apprécie au cas par cas selon plusieurs critères : la cible du produit, l’objectif du traitement ainsi que le degré de transformation de la denrée d’origine végétale à protéger. Un tableau visant à préciser la frontière entre les produits phytopharmaceutiques et les produits biocides a été mis en ligne sur le site du ministère chargé de l’environnement pour faciliter cet examen : http://www.developpementdurable. gouv.fr/IMG/pdf/Frontiere_Phyto-Biocides.pdf
IX. Exigences relatives à la distribution, le conseil ou l’application des produits phytopharmaceutiques
IX.1 Un agrément pour toutes les entreprises de distribution, d’application en prestation de service ou de conseil à l’utilisation des produits phytopharmaceutiques
Entre 1992 et fin octobre 2011, seuls les distributeurs de produits phytopharmaceutiques classés dans certaines catégories de danger (15) et les entreprises d’application en prestation de service étaient soumis à l’obtention d’un agrément et à la tenue d’un registre des ventes ou d’application de produits phytopharmaceutiques. Ces exigences étaient purement nationales.
Cet agrément était délivré par les directions régionales de l’alimentation, de l’agriculture et de la forêt, sous réserve de la justification :
1. de la souscription d’une police d’assurance couvrant la responsabilité civile
2. de l’emploi permanent pour les tâches d’encadrement et de formation liées à l’activité de distribution des produits phytopharmaceutiques de personnes qualifiées en effectif suffisant compte tenu de l’activité (doctrine : 1 employé sur 10 devait être titulaire du diplôme pour l’application des produits antiparasitaires – DAPA)
Le registre comportait pour tous les produits : le nom homologué, le numéro d’AMM et la quantité vendue, le montant de la redevance pour pollution diffuse le cas échéant. Pour tous les produits ne bénéficiant pas de la mention « EAJ », le registre comportait en plus, le numéro et la date de la facture, le code postal de l’utilisateur.
En application de la directive 2009/128/CE (16), cet agrément professionnel a été réformé en profondeur en 2011. Le champ de l’agrément a ainsi été élargi à tous les distributeurs (17) de produits phytopharmaceutiques, quel que soit le classement de danger, toutes les entreprises d’application en prestation de service et toutes les entreprises de conseil (18) à l’utilisation des produits phytopharmaceutiques (L.254-1 du code rural et de la pêche maritime).
(15) Ancien L.254-1 du code rural : « sont subordonnées à la détention d’un agrément et à la tenue d’un registre la mise en vente, la vente ou la distribution à titre gratuit aux utilisateurs des produits à usage agricole et des produits assimilés mentionnés à l’article L.253-+1 et classés, à l’issue de la procédure d’autorisation de mise sur le marché prévue aux articles L.253-1 à L.253-8 et L.253-14 à L.253-17 dans les catégories toxique, très toxique, cancérigène, mutagène, tératogène et dangereuse pour l’environnement. »
(16) Directive n° 2009/128/CE du Parlement européen et du Conseil du 21 octobre 2009 instaurant un cadre d’action communautaire pour parvenir à une utilisation des pesticides compatible avec le développement durable, transposée par le décret n°2011-1325 du 20 octobre 2011 fixant les conditions de délivrance, de renouvellement, de suspension et de retrait des agréments des entreprises et des certificats individuels pour la mise en vente, la distribution à titre gratuit, l’application et le conseil à l’utilisation des produits phytopharmaceutiques.
(17) Il s’agit des distributeurs de produits phytopharmaceutiques à des utilisateurs finaux : les premiers metteurs sur le marché et les grossistes ne sont pas concernés par cet agrément. En revanche, ils sont concernés par la certification des personnes imposée par la directive : voir plus loin.
(18) Chambres d’agriculture, conseillers privés.
IX.1.1 Les conditions relatives à l’agrément (R.254-15 à R.254-19 du code rural et de la pêche maritime)
En application de l’article L.254-2 du code rural et de la pêche maritime, l’agrément est délivré par le Préfet (19) de la région où se situe le siège social de l’entreprise, sous réserve de justifier :
1. de la souscription d’une police d’assurance couvrant la responsabilité civile professionnelle ;
2. de la certification de l’entreprise par un organisme certificateur accrédité (voir IX.1.2) ; 3. d’un contrat prévoyant le suivi nécessaire au maintien de la certification.
L’article R.254-17 prévoit qu’un numéro d’agrément est notifié à l’issue de l’instruction au demandeur dans un délai de deux mois. En cas de refus d’agrément, les motifs du refus sont également communiqués.
L’agrément est valable sans limitation de durée sous réserve que les conditions de l’agrément soient remplies. Chaque année, le détenteur de l’agrément fournit au préfet de région une copie de l’attestation de la souscription à une police d’assurance couvrant sa responsabilité civile professionnelle (R.254-19 du code rural et de la pêche maritime).
Par ailleurs, il faut signaler qu’en application de l’article R.254-27 du code rural et de la pêche maritime, le Préfet peut suspendre l’exercice de la ou des activités du détenteur de l'agrément dans tout ou partie des établissements du détenteur de l'agrément, s'il apparaît, lors de contrôles effectués par les agents CCRF que les conditions d’exercice ne sont pas satisfaites.
Enfin, en application de l’article L.254-6 du code rural et de la pêche maritime, les personnes qui exercent les activités soumises à agrément doivent faire référence dans leurs documents commerciaux à cet agrément et procéder à leur affichage dans les locaux accessibles à la clientèle. Elles doivent également tenir un registre de leurs ventes (voir IX.3).
(19) La direction régionale de l’alimentation, de l’agriculture et de la forêt de la région du siège social de l’entreprise est le service instructeur du dossier d’agrément.
IX.1.2 La certification d’entreprise : préalable à l’agrément (R.254-3 à R.254-7 du code rural et de la pêche maritime)
La certification d’entreprise est un préalable à l’obtention de l’agrément. Elle est obtenue à l’issue d’un audit réalisé par un organisme certificateur accrédité. La liste des organismes certificateurs reconnus est publiée sur le site internet du ministère chargé de l’agriculture. Le dispositif de certification est détaillé dans l’arrêté du 25 novembre 2011 fixant les modalités de la certification mentionnée au 2° de l’article L. 254-2 du code rural et de la pêche maritime.
Les entreprises entrant dans le champ de l’agrément doivent respecter :
- un référentiel commun à toutes les entreprises soumises à agrément, appelé « organisation générale », qui impose notamment le descriptif de l'organisation de l'entreprise et de ses différents sites (organigrammes fonctionnels, liste des personnels soumis à certificat individuel) et de la gestion des compétences. Ce référentiel a été publié par l’arrêté du 25 novembre 2011 relatif au référentiel de certification prévu à l’article R. 254-3 du code rural et de la pêche maritime « organisation générale ». Une des dispositions prévoit ainsi que toutes les personnes impliquées dans le champ des activités agréées doivent détenir un certificat individuel correspondant à leur fonction en cours de validité (voir IX.2).
- un référentiel d'activité qui décrit les différentes exigences pour l'activité, comme la traçabilité et le suivi de la mise en oeuvre de l'activité, le stockage et le transport des produits phytopharmaceutiques. Il existe 4 référentiels d’activité :
→ distribution de produits phytopharmaceutiques à des utilisateurs non professionnels (arrêté du 25 novembre 2011 relatif au référentiel de certification prévu à l’article R. 254-3 du code rural et de la pêche maritime pour l’activité « distribution de produits phytopharmaceutiques à des utilisateurs non professionnels ») ; une des nouveautés consiste à exiger, en application de l’article 6 de la directive 2009/128/CE, la disponibilité au moment de la vente d’une personne certifiée pour fournir au client les informations appropriées quant à l’utilisation des produits phytopharmaceutiques
→ distribution de produits phytopharmaceutiques à des utilisateurs professionnels (arrêté du 25 novembre 2011 relatif au référentiel de certification prévu à l’article R. 254-3 du code rural et de la pêche maritime pour l’activité « distribution de produits phytopharmaceutiques à des utilisateurs professionnels ») ; une des nouveautés consiste à exiger, comme pour la distribution à des utilisateurs non professionnels, la disponibilité au moment de la vente d’une personne certifiée pour fournir au client les informations appropriées quant à l’utilisation des produits phytopharmaceutiques.
→ application de produits phytopharmaceutiques en prestation de services (arrêté du 25 novembre 2011 relatif au référentiel de certification prévu à l’article R. 254-3 du code rural et de la pêche maritime pour l’activité « application en prestation de service de produits phytopharmaceutiques ») ;
→ conseil indépendant des activités de vente et d’application (arrêté du 25 novembre 2011 relatif au référentiel de certification prévu à l’article R. 254-3 du code rural et de la pêche maritime pour l’activité « conseil indépendant de toute activité de vente ou d’application »). Il doit y avoir une traçabilité écrite du conseil qui en précise les motivations. Des solutions alternatives de lutte contre les organismes nuisibles devront être proposées lorsqu’elles existent.
L’article L.254-7 prévoit que la préconisation écrite doit préciser la substance active, la spécialité recommandée, la cible, les parcelles concernées, la superficie à traiter, la dose recommandée et les conditions de mise en oeuvre.
Lors de la constatation d’un écart critique par rapport aux exigences du référentiel, l’organisme certificateur demande une mise en conformité pour corriger cet écart (sous un mois). La persistance de l’écart aboutit à la suspension de la certification. Toute suspension ou retrait de certification est notifiée au préfet de région (R.254-5 du code rural et de la pêche maritime).
IX.1.3 un basculement progressif entre l’ancien et le nouvel agrément jusqu’en octobre 2013 (I et II de l’article 3 du décret n° 2011-1325 du 18 octobre 2011 – non codifié)
Des dispositions transitoires permettent le basculement progressif dans le nouveau dispositif.
1. Concernant les entreprises de distribution et d’application en prestation de service qui sont déjà agréées (sur la base des anciennes conditions) :
→ jusqu'au 30 septembre 2012, elles sont assurées (assurance responsabilité civile professionnelle) et elles disposent d'une personne certifiée sur 10 personnes concernées par l’activité,
→ à partir du 1er octobre 2012, s'ajoute aux deux conditions précédentes la signature d'un contrat avec un organisme certificateur,
→ à partir du 1er octobre 2013, les entreprises doivent avoir obtenu leur certification délivrée par l'organisme certificateur, ce qui implique de respecter complètement les référentiels et d'avoir toutes les personnes concernées détentrices d'un certificat individuel.
2. Concernant les entreprises de conseil à l'utilisation des produits phytopharmaceutiques :
→ jusqu'au 30 septembre 2012, elles devront fournir leur attestation d'assurance,
→ à partir du 1er octobre 2012, s'ajoutent la signature d'un contrat avec un organisme certificateur et le fait de disposer d'un conseiller sur 3, titulaire d'un certificat individuel « conseil »,
→ à partir du 1er octobre 2013, ces entreprises doivent avoir obtenu leur certification délivrée par un organisme certificateur. Cela implique de respecter complètement les référentiels « conseil » et « organisation générale », et, en particulier, que tous les conseillers soient titulaires de leur certificat individuel.
3. Pour les entreprises de distribution qui entrent dans le champ de l’agrément (distributeurs de produits phytopharmaceutiques non classés ou classés dans d’autres catégories telles que nocif, irritant…) :
→ jusqu’au 30 septembre 2012, elles devront fournir leur attestation d’assurance,
→ à partir du 1er octobre 2012, s’ajoutent la signature d’un contrat avec un organisme certificateur et le fait de disposer d’une personne certifiée sur 3 concernées par l’activité,
→ à partir du 1er octobre 2013, ces entreprises doivent avoir obtenu leur certification délivrée par l’organisme certificateur. Cela implique de respecter complètement les référentiels « distribution de produits à usage non professionnel » et « organisation générale », et en particulier que la totalité des personnes concernées soient titulaires de leur certificat individuel.
IX.2 La certification individuelle des professionnels
L’article 5 de la directive n° 2009/128/CE prévoit que chaque Etat membre mette en place des systèmes de formation tant initiale que continue à l’intention des distributeurs (20), conseillers et utilisateurs professionnels (21) de produits phytopharmaceutiques ainsi que des systèmes de certification de ces professionnels. L’objectif est que ces professionnels soient parfaitement conscients des risques que ces produits présentent pour la santé humaine et pour l’environnement et soient pleinement informés des mesures à prendre pour réduire ces risques autant que possible.
Ainsi, l’article R.254-1 du code rural et de la pêche maritime introduit le « certificat individuel » qui est nécessaire à l’exercice des fonctions d’encadrement, de vente, de conseil et d’application en prestation de service ou pour son propre compte de produits phytopharmaceutiques.
(20) La notion de distributeur au sens de la directive 2009/128/CE regroupe toute personne physique ou morale qui met un produit phytopharmaceutique sur le marché, notamment les grossistes, détaillants, vendeurs et fournisseurs.
(21) La notion d’utilisateur professionnel au sens de l’article 3 de la directive 2009/128 correspond à toute personne qui utilise des pesticides au cours de son activité professionnelle, et notamment les opérateurs, les techniciens, les employeurs et les indépendants, tant dans le secteur agricole que dans d’autres secteurs. Cela englobe notamment tous les agriculteurs et les personnels des collectivités territoriales.
IX.2.1 Les conditions relatives à la certification individuelle (R.254-8 à R.254-14 du code rural et de la pêche maritime)
Plusieurs certificats individuels ont été définis selon l’activité du professionnel. Ils peuvent être obtenus par la formation et / ou un test de connaissance.
Pour les utilisateurs professionnels, les certificats sont adaptés selon :
- La fonction exercée quant à l’utilisation des produits : fonction de décision (décideur) ou d’exécution (opérateur),
- Le lieu d’activité : hors exploitation agricole ou en exploitation agricole.
Pour les distributeurs, une différenciation est opérée entre les produits : un certificat pour la vente et la distribution des produits professionnels et un certificat pour la vente et la distribution des produits grand public.
Pour les personnes exerçant une activité de conseil, il est créé un certificat « conseil à l'utilisation des produits phytopharmaceutiques ».
Les formations sont dispensées par des organismes habilités et répertoriés par les directions régionales de l’alimentation, de l’agriculture et de la forêt.
Les certificats individuels sont délivrés par la direction régionale de l’alimentation, de l’agriculture et de la forêt de la région du domicile du demandeur, pour une durée de cinq ans renouvelable, portée à 10 ans pour ceux permettant l’utilisation des produits phytopharmaceutiques dans le cadre d’une activité agricole.
Jusqu'au 31 décembre 2011, les personnes exerçant les activités de distribution ou d'application en prestation de service des produits phytopharmaceutiques pouvaient solliciter la délivrance d'un certificat soit dans les conditions antérieures (22) à la date de publication du décret n° 2011-1325, soit dans les conditions prévues par les articles R. 254-8 à R. 254-14 du code rural et de la pêche maritime dans leur rédaction issue de ce décret.
A partir du 1er octobre 2014, les applicateurs professionnels agissant à titre salarié, pour leur propre compte ou dans le cadre d’un contrat d’entraide à titre gratuit (II du L.254-3 du code rural et de la pêche maritime), ainsi que les personnes qui mettent des produits phytopharmaceutiques sur le marché autres que celles soumises à agrément (23) (IV du L.254-1 du code rural et de la pêche maritime) justifient de l’obtention d’un certificat individuel.
Jusqu’au 26 novembre 2015, le certificat est également attribué aux professionnels, ressortissants d'un Etat membre de l'Union européenne ou d'un autre Etat partie à l'accord sur l'Espace économique européen, sous réserve qu’ils justifient d’un titre, d’un diplôme, d’une attestation de connaissance ou de deux ans d’expérience si cette activité n’est pas encore réglementée dans cet Etat.
(22) Il s’agit des diplômes dits DAPA (diplôme d’applicateurs de produits antiparasitaires) et des certiphytos 2009-2010.
(23) c'est-à-dire les grossistes et les fournisseurs
IX.3 Le registre d’activité (R.254-23 à R.254-26 du code rural et de la pêche maritime)
En application de l’article L.254-6 du code rural et de la pêche maritime, les personnes qui exercent des activités soumises à agrément (application en prestation de service, distribution à des utilisateurs et conseil à l’utilisation) tiennent, de façon méthodique et chronologique, un registre de leur activité : registre des ventes ou registre de l’utilisation.
→ Le registre des ventes
Dans le cas de la distribution de produits phytopharmaceutiques à des utilisateurs ou de la distribution de semences traitées par un produit phytopharmaceutique aux utilisateurs de ces semences, il s’agit d’un registre des ventes.
L’article R.254-23 du code rural et de la pêche maritime prévoit que le registre des ventes comporte :
1. Pour tous les produits (professionnels et grand public) :
- le nom commercial du produit ;
- le numéro d'autorisation de mise sur le marché ;
- la quantité vendue ou distribuée ;
- le montant de la redevance correspondant à cette quantité.
2. En outre, pour les produits (24) destinés aux professionnels :
- le numéro de facture et la date de facturation, s'il y a lieu ;
- le code postal de l'utilisateur final ;
- les références attestant de sa qualité d'utilisateur professionnel (voir IX.4). Il s’agit d’une nouveauté introduite par le décret n° 2011-1650 du 25 novembre 2011.
3. Pour toutes les semences traitées :
- l'espèce végétale de la semence traitée ou, dans le cas des mélanges de semences pour gazon, la mention " gazon " ;
- la quantité vendue, en quintal ou en nombre de milliers de grains ;
- pour chaque produit utilisé pour traiter cette semence : le nom commercial du produit, son numéro d’autorisation de mise sur le marché, la quantité du produit correspondant à la quantité de semences vendues, le montant de la redevance correspondant à la quantité de semences vendues.
Toutes ces mentions sont portées au registre dans un délai de deux mois à compter de la vente ou la distribution du produit (R.254-24 du code rural et de la pêche maritime).
En outre, le registre des ventes comprend en annexe le bilan nécessaire à la déclaration de la redevance pour pollutions diffuses (25).
→ Le registre d’utilisation
Dans le cas de l’activité de traitement de semences en prestation de service soumise à agrément, il s’agit d’un registre de l’utilisation des produits phytopharmaceutiques.
L’article R.254-23-1 du code rural et de la pêche maritime prévoit que ce registre d’utilisation comporte pour chaque établissement et pour chaque produit phytopharmaceutique utilisé pour le traitement des semences pour le compte de leur utilisateur final quand ce dernier n'est pas une personne produisant des semences en vue de leur mise sur le marché, les indications suivantes :
- la date du traitement ;
- le numéro de facture et la date de facturation ;
- le code postal du commanditaire pour le compte duquel est réalisé le traitement de semence en prestation de service ;
- le nom commercial et le numéro d'autorisation de mise sur le marché du produit utilisé pour ce traitement ;
- l'espèce végétale ou, dans le cas des mélanges de semences pour gazon, la mention " gazon " et la quantité de semences traitées au moyen de ce produit ;
- la quantité du produit utilisée ;
- le numéro d'agrément de l'établissement d'achat du produit, ou à défaut, son numéro d'identification SIRET ou, en l'absence de ce numéro, sa localisation géographique ;
- le montant de redevance correspondant à la quantité de produit utilisée.
En outre, le registre d’utilisation comprend en annexe le bilan nécessaire à la déclaration de la redevance pour pollutions diffuses (26).
Par ailleurs, il faut préciser qu’il existe un troisième type de registre : le registre d’achat. En application de l’article L.254-3-1 du code rural et de la pêche maritime, il concerne toute personne qui, dans le cadre d'une activité professionnelle (autre que la prestation de service de traitement de semences) acquiert, à titre onéreux ou gratuit, en vue de son utilisation un produit phytopharmaceutique ou une semence traitée ou commande une prestation de traitement de semence au moyen de ces produits auprès d'une personne qui n'est pas redevable de la redevance pour pollutions diffuses (exemple : un agriculteur français qui achète un produit phytopharmaceutique à l’étranger). Cette personne doit alors inscrire dans un registre établi à cet effet le montant et la date de l'acquisition des produits ou de la prestation de traitement ainsi que les quantités de produits correspondantes.
L’article R.254-23-2 du code rural et de la pêche maritime prévoit que ce registre d’achat comporte :
1. Pour chaque produit acheté auprès d'une personne qui n'est pas redevable de la redevance pour pollutions diffuses :
- le numéro de facture et la date de facturation ;
- le nom commercial du produit et son numéro d'autorisation de mise sur le marché à l'étranger ;
- le nom du produit français de référence et son numéro d'autorisation de mise sur le marché en France ;
- la quantité achetée ;
- le montant de l'achat ;
- le montant de redevance correspondant à cette quantité ;
2. Pour chaque semence traitée acquise auprès d'une personne qui n'est pas redevable de la redevance pour pollutions diffuses ou dont le traitement a été réalisé par un prestataire de
service qui n'est pas redevable de la redevance pour pollutions diffuses :
- le numéro de facture et la date de facturation ;
- l'espèce végétale de la semence ou, dans le cas des mélanges de semences pour gazon, la mention " gazon " ;
- la quantité de semence acquise ou traitée, en quintal ;
- le montant de l'achat ou de la prestation de service ;
- pour chaque produit utilisé pour traiter cette semence : le nom commercial du produit et le numéro d'autorisation de mise sur le marché à l'étranger ; le nom du produit de référence et son numéro d'autorisation de mise sur le marché en France ; la quantité de ce produit correspondant à la quantité de semence ; le montant de redevance correspondant à cette quantité.
Ce registre peut être constitué des factures d'achat de produit, de semences traitées ou de prestation de traitement de semences à condition que ces factures recensent toutes les mentions précédemment citées.
Toutes ces mentions sont portées au registre dans un délai de deux mois à compter de la vente ou la distribution du produit (R.254-24 du code rural et de la pêche maritime).
En outre, le registre d’achat comprend en annexe le bilan nécessaire à la déclaration de la redevance pour pollutions diffuses.
(24) dont l'autorisation ne porte pas la mention " emploi autorisé dans les jardins »
(25) En application de l’article L.213-10-8 du code de l’environnement, toute personne qui, dans le cadre d'une activité professionnelle, exceptés le traitement en prestation de service et la distribution de semences traitées, acquiert un produit phytopharmaceutique ou une semence traitée au moyen de ces produits ou commande une prestation de traitement de semence au moyen de ces produits est assujettie à une redevance pour pollutions diffuses. Les distributeurs font apparaître le montant de la redevance qu'ils ont acquittée au titre du produit distribué sur leurs factures, à l'exception des produits distribués portant la mention « emploi autorisé dans les jardins ». Les modalités de déclaration et de reversement aux agences et offices de l’eau de la redevance pour pollutions diffuses sont énoncées à l’article R.213-48-13 du code de l’environnement
(26) En application de l’article L.213-10-8 du code de l’environnement, le prestataire de traitement de semences n’est pas assujetti à la redevance pour pollution diffuses mais son client l’est.
IX.4 La vente des produits phytopharmaceutiques destinés aux utilisateurs professionnels
L’article R. 254-20 du code rural et de la pêche maritime, en application de l’article 6 de la directive 2009/128/CE impose la restriction de vente de produits professionnels aux seuls titulaires du certificat individuel (produits professionnels réservés aux utilisateurs professionnels).
Ainsi les distributeurs doivent s’assurer, préalablement à la vente des produits ne portant pas la mention « emploi autorisé dans les jardins », de la qualité d’utilisateur professionnel de l’acheteur, sur présentation par celui - ci de justificatifs. Ces derniers sont nombreux et énoncés dans l’arrêté du 30 décembre 2010 relatif aux références exigées des utilisateurs professionnels de produits phytopharmaceutiques. Toutefois, à compter du 1er janvier 2015, un seul justificatif sera accepté : il s’agira du numéro du certificat individuel de l’acheteur.
IX.5 Les règles générales d’utilisation des produits phytopharmaceutiques
L’article 55 du RCE n°1107/2009 exige que les produits phytopharmaceutiques fassent l’objet d’une utilisation appropriée. Celle-ci inclut l’application des principes de bonnes pratiques phytosanitaires et le respect des conditions fixées dans l’autorisation de mise sur le marché et mentionnées sur l’étiquetage. Elle est en outre conforme aux dispositions de la directive 2009/128/CE, et en particulier aux principes généraux de lutte intégrée contre les ennemis des cultures.
En outre, le projet de décret relatif à la mise sur le marché et l’utilisation des produits phytopharmaceutiques prévoit que les personnes souhaitant introduire, pour leur usage personnel, un produit phytopharmaceutique pour lequel un permis de commerce parallèle a déjà été délivré, déclarent au préfet de la région du lieu de leur résidence administrative, les quantités nécessaires à cet usage et la date d'introduction des produits, dans un délai minimum de 15 jours ouvrés avant cette date. De plus, cette personne, lorsqu’elle exerce une activité agricole, devra afficher dans son local de stockage des produits phytopharmaceutiques, les mentions d’étiquetage obligatoire liées à ce produit.
Enfin, il est nécessaire de rappeler l’existence d’arrêtés nationaux encadrant l’utilisation des produits phytopharmaceutiques :
- L’arrêté du 12 septembre 2006 relatif à la mise sur le marché et à l’utilisation des produits visés à l’article L. 253-1 du code rural ;
- L’arrêté du 7 avril 2010 relatif à l’utilisation des mélanges extemporanés de produits visés à l’article L. 253-1 du code rural ;
- L’arrêté du 31 mai 2011 relatif aux conditions d’épandage des produits mentionnés à l’article L. 253-1 du code rural et de la pêche maritime par voie aérienne (arrêté transposant l’article 9 de la directive 2009/128/CE) ;
- L’arrêté du 27 juin 2011 relatif à l’interdiction d’utilisation de certains produits mentionnés à l’article L. 253-1 du code rural et de la pêche maritime dans des lieux fréquentés par le grand public ou des groupes de personnes vulnérables (arrêté transposant l’article 12 de la directive 2009/128/CE) ;
IX.6 L’utilisation des produits phytopharmaceutiques en agriculture biologique
L’agriculture biologique est un système de production agricole spécifique dans lequel l’utilisation d’intrants, et plus particulièrement ceux issus de la chimie de synthèse, est restreint.
La lutte contre les parasites et maladies en agriculture biologique est prioritairement conduite par les mesures détaillées à l’article 12 (27) du Règlement (CE) n° 834/2007 du Conseil du 28 juin 2007 relatif à la production biologique et à l'étiquetage des produits biologiques et abrogeant le règlement (CEE) n° 2092/91. Celles-ci correspondent principalement à la mise en place de pratiques culturales et des pratiques du sol compatibles avec ce mode de production comme le recours à des espèces et des variétés appropriées, la mise en place de rotations adaptées des cultures et le maintien des ennemis
naturels des ravageurs.
Lorsque ces mesures ne suffisent pas à protéger les végétaux contre les ravageurs et les maladies, l’utilisation de produits phytopharmaceutiques est autorisée en application de l’article 5 du règlement (CE) n° 834/2007. Toutefois, seuls les produits dont les substances actives sont listées à l’annexe II du Règlement (CE) n° 889/2008 (28) peuvent être utilisés. Dans ce cadre, les utilisateurs conservent des documents justificatifs attestant la nécessité de recourir à l’emploi de ces produits.
Un guide des intrants utilisables en agriculture biologique en France a été publié sur le site internet de l’INAO (Institut national de l’origine et de la qualité) en juin 2011 : http://www.inao.gouv.fr/repository/editeur/pdf/GUIDES_et_NT/111012_GUID…
Il recense les spécialités commerciales bénéficiant d’une autorisation de mise sur le marché national dont les substances actives sont listées à l’annexe II du règlement (CE) n° 889/2008.
En outre, il est précisé que l’apposition de la mention « utilisable en agriculture biologique » sur l’étiquetage des produits phytopharmaceutiques relève d’une démarche volontaire du responsable de la mise sur le marché qui entend ainsi n’introduire dans son produit que des substances actives listées à l’annexe II du règlement (CE) n° 889/2008. Il est rappelé que ce règlement ne fixe aucune restriction quant à la nature des coformulants incorporés dans les produits phytopharmaceutiques.
(27) paragraphe 1 ; points a), b), c), g) et h)
(28) Règlement (CE) n° 889/2008 de la Commission du 5 septembre 2008 portant modalités d’application du règlement (CE) n°834/2007 du Conseil relatif à la production biologique et à l’étiquetage des produits biologiques en ce qui concerne la production biologique, l’étiquetage et les contrôles
X. Habilitations et pouvoirs d’enquête des agents CCRF pour le contrôle de la mise sur le marché et de l’utilisation des produits phytopharmaceutiques
Le contrôle de la mise sur le marché et de l’utilisation des produits phytopharmaceutiques est une obligation qui résulte de l’article 68 du RCE n° 1107/2009.
Les agents de la concurrence, de la consommation et de la répression des fraudes sont habilités par les articles L.253-14 et L.254-11 du code rural et de la pêche maritime, à rechercher et constater les infractions aux chapitres III et IV du titre V du livre II du même code et aux textes pris pour leur application.
Les pouvoirs d’enquête sont ceux prévus aux articles R.215-1 et suivants du code de la consommation.
Il est à noter que, suite à une erreur matérielle, le chapitre V du titre Ier du livre II du code de la consommation n’est actuellement pas cité aux articles L.253-14 et L.254-11 du code rural et de la pêche maritime. Un correctif sera prochainement apporté afin de citer explicitement ce chapitre V dans les articles L.253-14 et L.254-11 du code rural et de la pêche maritime.
XI. Tableau des principales infractions et sanctions relatives à la mise sur le marché et l’utilisation des produits phytopharmaceutiques
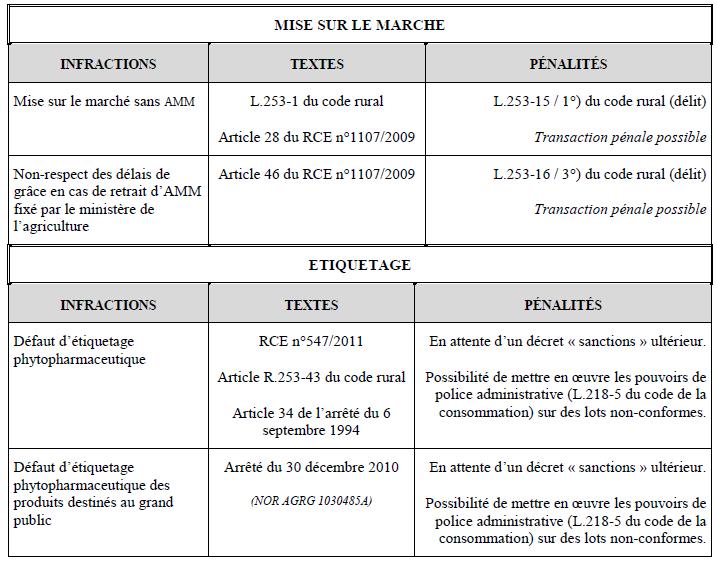
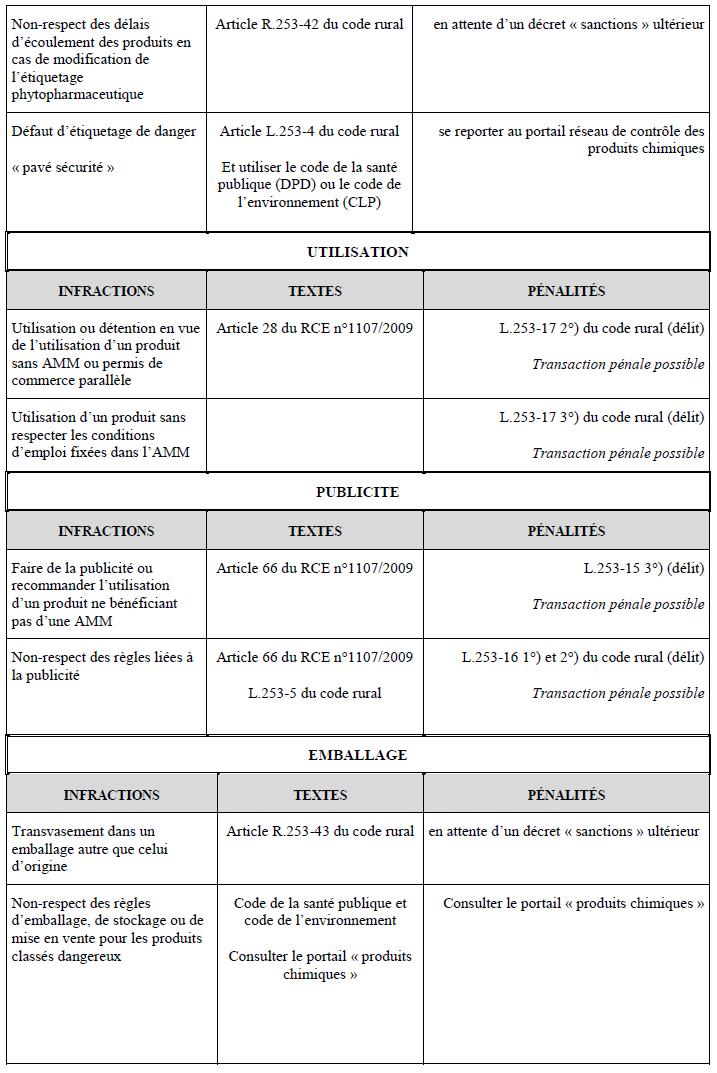
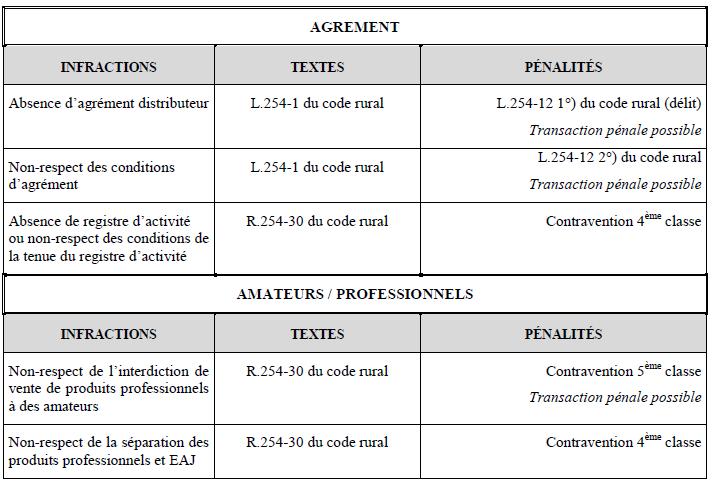
Il est précisé qu’en application des articles L.205-10 et R.205-3 à R.205-5 du code rural et de la pêche maritime, l’autorité administrative peut, tant que l’action publique n’a pas été mise en mouvement et après avoir recueilli l’accord du Procureur de la République, transiger sur la poursuite des contraventions (5ème classe uniquement) et délits prévus pour le contrôle de la mise sur le marché des produits phytopharmaceutiques.